Chapter: Clinical Anesthesiology: Perioperative & Critical Care Medicine: Acid-Base Management
Respiratory Acidosis
RESPIRATORY ACIDOSIS
Respiratory acidosis is defined as a primary increase in Paco2. This
increase drives the reaction
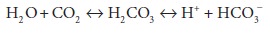
to the right, leading to an increase in [H+] and a decrease in arterial pH. For the reasons described above, [HCO3−] is minimally affected.
Paco2 represents the balance between CO2 pro-duction and CO2
elimination:
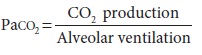
CO2 is a byproduct of fat and carbohydrate metabolism. Muscle activity,
body temperature, and thyroid hormone activity can all have major influ-ences
on CO2
production. Because CO 2 production does not appreciably vary under most circum-stances,
respiratory acidosis is usually the result of alveolar hypoventilation (Table
50–3). In patients
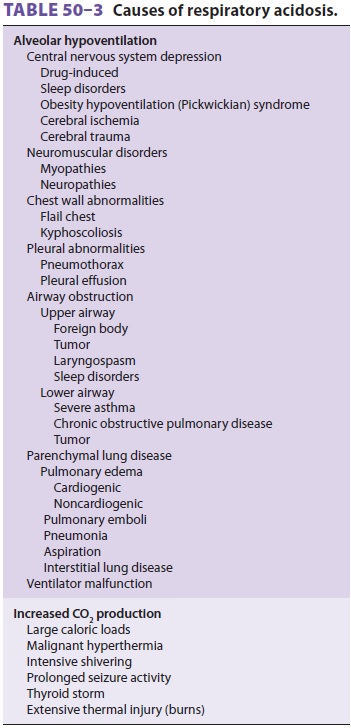
with a limited capacity to increase alveolar ventila-tion, however,
increased CO 2 production can pre-cipitate respiratory acidosis.
Acute Respiratory Acidosis
The compensatory response to acute (6–12 h) elevations in Paco2 is
limited. Buffering is primar-ily provided by hemoglobin and the exchange of
extracellular H+ for Na+ and K+ from bone and the intracellular fluid compartment (see above). The
renal response to retain more bicarbonate is acutely very limited. As a result,
plasma [HCO3−] increases only about 1 mEq/L
for each 10 mm Hg increase in Paco2 above 40 mm Hg.
Chronic Respiratory Acidosis
“Full” renal compensation characterizes
chronic respiratory acidosis. Renal compensation is appre-ciable only after
12–24 hr
and may not peak until 3–5 days. During that
time, the sustained increase in Paco2 has been present long enough to permit maximal renal
compensation. During chronic respiratory acidosis, plasma
[HCO 3−] increases approximately 4
mEq/L for each 10 mm Hg increase in Paco2 above 40 mm Hg.
Treatment of Respiratory Acidosis
Respiratory acidosis is treated by reversing the
imbalance between CO 2 production and alveolar ventilation. In most instances,
this is accomplished by increasing alveolar ventilation. Measures aimed at
reducing CO2 production are useful only in specific instances (eg, dantrolene
for malignant hyperther-mia, muscle paralysis for tetanus, antithyroid
medi-cation for thyroid storm, and reduced caloric intake in patients receiving
enteral or parenteral nutrition). Potential temporizing measures aimed at
improv-ing alveolar ventilation (in addition to controlled mechanical
ventilation) include bronchodilation, reversal of narcosis, or improving lung
compliance (diuresis). Severe acidosis (pH <7.20), CO2 narcosis, and
respiratory muscle fatigue are indications for mechanical ventilation. An
increased inspired oxy-gen concentration is also usually necessary, as
coex-istent hypoxemia is common. Intravenous NaHCO3 is rarely necessary, unless
pH is <7.10 and HCO3− is <15 mEq/L. Sodium bicarbonate therapy will
tran-siently increase Paco2:
H+
+ HCO3−↔ CO2+ H2O
Buffers that do not produce CO 2, such
as CarbicarbTM or tromethamine (THAM), are theoret-ically attractive
alternatives; however, there is almost no evidence showing that they have
greater efficacy than bicarbonate. Carbicarb TM is a mixture of 0.3
M sodium bicarbonate and 0.3 M sodium carbonate;
buffering by this mixture mainly produces sodium
bicarbonate instead of CO2. Tromethamine has the added advantage of
lacking sodium and may be a more effective intracellular buffer.
Patients with a baseline chronic respiratory
acidosis require special consideration. When such patients develop acute ventilatory
failure, the goal of therapy should be to return Paco2 to the
patient’s “normal” baseline. Normalizing the patient’s Paco2 to 40
mm Hg will produces the equivalent of a respi-ratory alkalosis . Oxygen therapy
must also be carefully controlled, because the respiratory drive in these
patients may be dependent on hypox-emia, not Paco2. “Normalization”
of Paco2 or relative hyperoxia can precipitate severe
hypoventilation.
Related Topics