Chapter: Clinical Anesthesiology: Perioperative & Critical Care Medicine: Acid-Base Management
Renal Acid-Base Compensation
RENAL COMPENSATION
The ability of the kidneys to control the
amount of HCO3− reabsorbed from filtered tubular fluid, form new HCO3−,
and eliminate H+
in the form of titratable acids and ammonium ions allows them to exert a major influence on pH
during both metabolic and respiratory acid–base disturbances. In fact, the
kidneys are responsible for eliminating the approximately 1 mEq/kg per day of
sulfuric acid, phosphoric acid, and incompletely oxidized organic acids that
are normally produced by the metabolism of dietary and endogenous proteins,
nucleoproteins, and organic phosphates (from phosphoproteins and
phospholipids). Metabolism of nucleoproteins also produces uric acid.
Incomplete combustion of fatty acids and glucose produces keto acids and lactic
acid. Endogenous alkali are produced during the metabo-lism of some anionic
amino acids (glutamate and aspartate) and other organic compounds (citrate,
acetate, and lactate), but the quantity is insufficient to offset the
endogenous acid production.
Renal Compensation During Acidosis
The renal response to acidemia is
3-fold:increased reabsorption of the filtered HCO3−, (2) increased excretion of
titratable acids, and (3) increased production of ammonia. Although these
mechanisms are probably activated immedi-ately, their effects are generally not
appreciable for 12–24 hr and may not be maximal for up to 5 days.
A.Increased Reabsorption of HCO3−
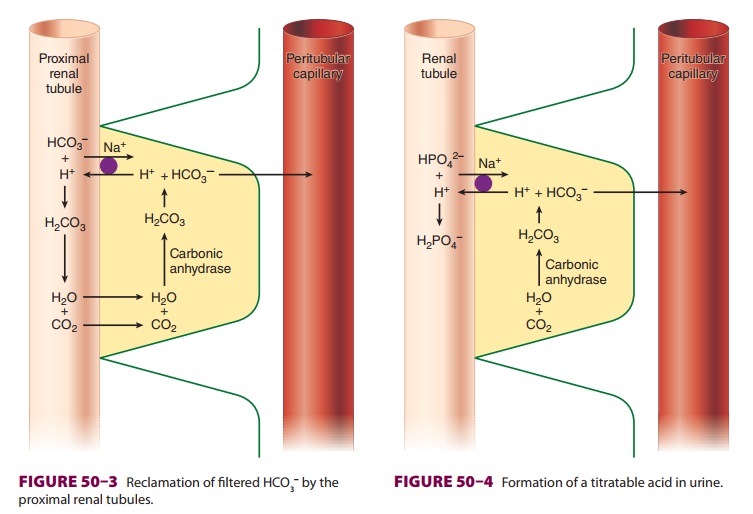
Bicarbonate reabsorption is shown in Figure 50–3. CO2 within
renal tubular cells combines with water in the presence of carbonic anhydrase.
The carbonic acid (H2CO3) formed rapidly dissociates into
H+ and HCO3−. Bicarbonate ion then enters the
blood-stream while the H + is secreted into the renal tubule, where it
reacts with filtered HCO3− to form H2CO3.
Carbonic
anhydrase associated with the luminal brush border catalyzes the dissociation
of H2CO3 into CO2 and H2O. The CO2
thus formed can dif-fuse back into the renal tubular cell to replace the CO2
originally consumed. The proximal tubules normally reabsorb 80% to 90% of the
filtered bicar-bonate load along with sodium, whereas the distal tubules are
responsible for the remaining 10% to 20%. Unlike the proximal H + pump, the H + pump in the distal tubule is not
necessarily linked to sodium reabsorption and is capable of generating steep H+ gradients between tubular fluid and
tubular cells. Urinary pH can decrease to as low as 4.4 (compared with a pH of
7.40 in plasma).
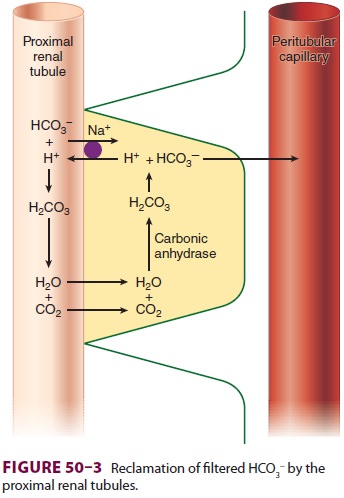
B. Increased Excretion of Titratable Acids
After all of the HCO3− in tubular fluid is
reclaimed, the H+ secreted into the tubular lumen can com-bine
with HPO42− to form H2PO4− (Figure 50–4); the latter is not
readily reabsorbed because of its charge and is eliminated in urine. The net
result
is that H+ is excreted from the body as H2PO4−, and the HCO3− that is generated in the process can
enter the bloodstream. With a pK of
6.8, the H2PO4−/HPO42− pair is normally an ideal urinary
buffer. When urinary pH approaches 4.4, however, all of the phosphate reaching
the distal tubule is in the H2PO4− form; HPO42− ions are no longer avail-able for
eliminating H+.
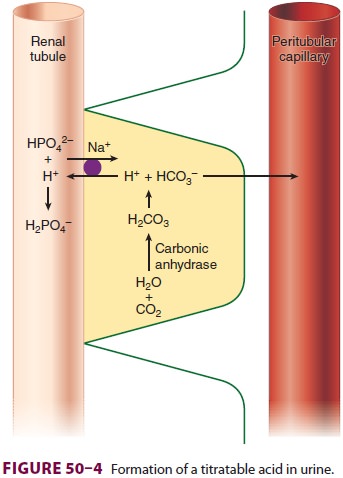
C. Increased Formation of Ammonia
After complete reabsorption of HCO3− and con-sumption of the
phosphate buffer, the NH3/NH4+ pair becomes the most
important urinary buffer (Figure
50–5). Deamination of glutamine within the mitochondria of proximal tubular
cells is the principal source of NH 3 production in the kidneys.
Acidemia markedly increases renal NH3 production. The ammonia formed
is then able to passively cross the cell’s luminal membrane, enter the tubular
fluid, and react with H+ to form NH4+. Unlike NH 3,
NH4+ does not readily penetrate the luminal membrane
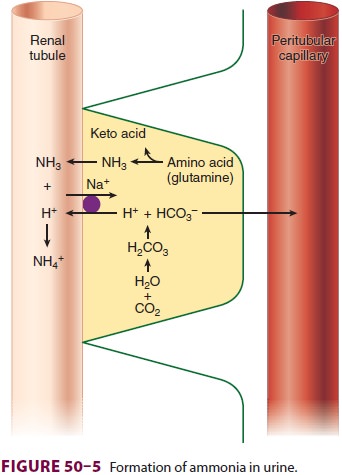
and is therefore trapped within the tubules. Thus,
excretion of NH 4+ in urine effectively eliminates H+.
Renal Compensation During Alkalosis
The tremendous amount of HCO3− normally filtered and subsequently reabsorbed allows the kidneys to rapidly excrete large amounts of bicarbonate, if necessary . As a result, the kidneys are highly effective in protecting against metabolic alkalosis, which therefore generally occurs only in association with concomitant sodium deficiency or mineralocorticoid excess. Sodium depletion decreases extracellular fluid volume and enhances Na+ reabsorption in the proximal tubule. To main-tain neutrality, the Na+ ion is brought across with a Cl− ion. As Cl − ions decrease in number (<10 mEq/L of urine), HCO3− must be reabsorbed. Increased H+ secretion in exchange for augmented Na + reabsorp-tion favors HCO 3− formation with metabolic alka-losis. Similarly, increased mineralocorticoid activity augments aldosterone-mediated Na+ reabsorption in exchange for H+ secretion in the distal tubules. Theresulting increase in HCO3− formation can initiate or propagate metabolic alkalosis. Metabolic alkalo-sis is commonly associated with increased mineralo-corticoid activity, even in the absence of sodium and chloride depletion.
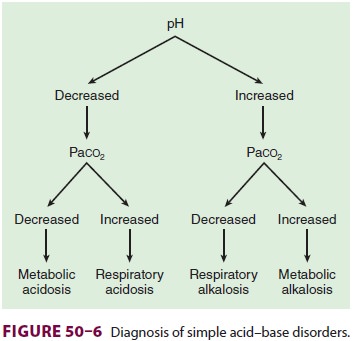
Base Excess
Base excess is the amount of acid or base
(expressed in mEq/L) that must be added for blood pH to return to 7.40 and Paco2 to return to 40 mm Hg at full O 2 saturation and 37°C. Moreover, it adjusts for noncarbonic
buffering in the blood. Simplistically, base excess represents the metabolic
component of an acid–base disturbance. A positive value indicates metabolic
alkalosis, whereas a negative value reveals metabolic acidosis. Base excess is
usually derived graphically or electronically from a nomogram originally
developed by Siggaard–Andersen and requires measurement of hemoglobin
concentration (Figure 50–6).
Related Topics