Chapter: Clinical Anesthesiology: Perioperative & Critical Care Medicine: Acid-Base Management
Metabolic Acidosis
METABOLIC ACIDOSIS
Metabolic acidosis is defined as a primary decrease in [HCO 3−]. Pathological processes can initiate metabolic acidosis by one of three mechanisms:
(1) consumption of HCO3− by a strong nonvolatile acid, (2) renal or gastrointestinal wasting of bicar-bonate, or (3) rapid dilution of the extracellular fluid compartment with a bicarbonate-free fluid.
A fall in plasma [HCO3−] without a propor-tionate reduction in Paco2 decreases arterial pH. The pulmonary compensatory response in a simple metabolic acidosis (see above) characteristically does not reduce Paco2 to a level that completely normalizes pH but can produce marked hyperven-tilation (Kussmaul’s respiration).
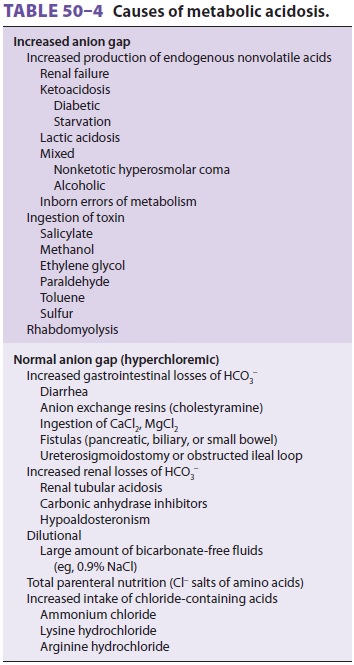
Table 50–4 lists disorders that can cause meta-bolic acidosis. Note that differential diagnosis of metabolic acidosis may be facilitated by calculation of the anion gap.
The Anion Gap
The anion gap in plasma is most commonly defined as the difference between the major measured cat-ions and the major measured anions:
Anion gap = major plasma cations - major plasma anions
Or
Anion gap = [Na+ ]− ([Cl− ]+[HCO3− ]
Some physicians include plasma K+ in the cal-culation. Using normal values,
Anion gap = 140 − (104 + 24) = 12 mEq/L x (normal range = 7 − 14 mEq/L)
In reality, an anion gap cannot exist because electroneutrality must be maintained in the body; the sum of all anions must equal the sum of all cat-ions. Therefore,
Anion gap = unmeasured anions - unmeasured cations
“Unmeasured cations” include K+, Ca2+, and Mg2+, whereas “unmeasured anions” include all organic anions (including plasma proteins), phos-phates, and sulfates. Plasma albumin normally accounts for the largest fraction of the anion gap (about 11 mEq/L). The anion gap decreases by 2.5 mEq/L for every 1 g/dL reduction in plasma albumin concentration. Any process that increases “unmeasured anions” or decreases “unmeasured cations” will increase the anion gap. Conversely, any process that decreases “unmeasured anions” or increases “unmeasured cations” will decrease the anion gap.
Mild elevations of plasma anion gap up to 20 mEq/L may not be helpful diagnostically dur-ing acidosis, but values >30 mEq/L usually indicate the presence of a high anion gap acidosis (below). Metabolic alkalosis can also produce a high anion gap because of extracellular volume depletion, an increased charge on albumin, and a compensatory increase in lactate production. A low plasma anion gap may be encountered with hypoalbuminemia, bro-mide or lithium intoxication, and multiple myeloma.
High Anion Gap Metabolic Acidosis
Metabolic acidosis with an increased anion gap is characterized by an increase in relatively strong nonvolatile acids. These acids dissociate into H+ and their respective anions; the H+ consumes HCO3− to produce CO2, whereas their anions (conjugate bases) accumulate and take the place of HCO3− in extracellu-lar fluid (hence the anion gap increases). Nonvolatile acids can be endogenously produced or ingested.
A. Failure to Excrete Endogenous Nonvolatile Acids
Endogenously produced organic acids are nor-mally eliminated by the kidneys in urine (above). Glomerular filtration rates below 20 mL/min (renal failure) typically result in progressive metabolic aci-dosis from the accumulation of these acids.
B. Increased Endogenous Nonvolatile Acid Production
Severe tissue hypoxia following hypoxemia, hypo-perfusion (ischemia), or an inability to utilize oxygen (cyanide poisoning) can result inlactic aci-dosis. Lactic acid is the end product of the anaerobic
metabolism of glucose (glycolysis) and can rapidly accumulate under these conditions. Decreased uti-lization of lactate by the liver, and, to a lesser extent by the kidneys, is less commonly responsible for lac-tic acidosis; causes include hypoperfusion, alcohol-ism, and liver disease. Lactate levels can be readily measured and are normally 0.3–1.3 mEq/L. Acidosis resulting from D-lactic acid, which is not recognized by α-lactate dehydrogenase (and not measured by routine assays), may be encountered in patients with short bowel syndromes; D-lactic acid is formed by colonic bacteria from dietary glucose and starch and is absorbed systemically.
An absolute or relative lack of insulin can result in hyperglycemia and progressive ketoacidosis from the accumulation of β-hydroxybutyric and aceto-acetic acids. Ketoacidosis may also be seen following starvation and alcoholic binges. The pathophysi-ology of the acidosis often associated with severe alcoholic intoxication and nonketotic hyperosmolar coma is complex and may represent a build-up of lactic, keto, or other unknown acids.
Some inborn errors of metabolism, such as maple syrup urine disease, methylmalonic aciduria, propionic acidemia, and isovaleric acidemia, pro-duce a high anion gap metabolic acidosis as a result of accumulation of abnormal amino acids.
C. Ingestion of Exogenous Nonvolatile Acids
Ingestion of large amounts of salicylates frequently results in metabolic acidosis. Salicylic acid and other acid intermediates rapidly accumulate and produce a high anion gap acidosis. Because salicylates also produce direct respiratory stimulation, most adults develop mixed metabolic acidosis with superim-posed respiratory alkalosis. Ingestion of methanol (methyl alcohol) frequently produces acidosis and visual disturbances (retinitis). Symptoms are typi-cally delayed until the slow oxidation of methanol by alcohol dehydrogenase produces formic acid, which is highly toxic to the retina. The high anion gap represents the accumulation of many organic acids, including acetic acid. The toxicity of ethylene glycol is also the result of the action of alcohol dehy-drogenase to produce glycolic acid. Glycolic acid, the principal cause of the acidosis, is further metab-olized to form oxalic acid, which can be deposited in the renal tubules and result in renal failure.
Normal Anion Gap Metabolic Acidosis
Metabolic acidosis associated with a normal anion gap is typically characterized by hyperchloremia. Plasma [Cl−] increases to take the place of the HCO3− ions that are lost. Hyperchloremic metabolic acidosis most commonly results from abnormal gastroin-testinal or renal losses of HCO3−, or from excessive intravenous administration of 0.9% NaCl solution.
Calculation of the anion gap in urine can be helpful in diagnosing a normal anion gap acidosis.
Urine anion gap = ([Na+] + [K+]) − [Cl−]
The urine anion gap is normally positive or close to zero. The principal unmeasured urinary cation is normally NH 4+, which should increase (along with Cl−) during a metabolic acidosis; the latter results in a negative urinary anion gap. Impairment of H+ or NH4+secretion, as occurs in renal failure or renal tubular acidosis (below), results in a positive urine anion gap in spite of systemic acidosis.
A. Increased Gastrointestinal Loss of HCO3−
Diarrhea is a common cause of hyperchloremic metabolic acidosis. Diarrheal fluid contains20–50 mEq/L of HCO 3−. Small bowel, biliary, and pancreatic fluids are all rich in HCO3−. Loss of large volumes of these fluids can lead to hyperchloremic metabolic acidosis. Patients with ureterosigmoidos-tomies and those with ileal loops that are too long or that become partially obstructed frequently develop hyperchloremic metabolic acidosis. The inges-tion of chloride-containing anion-exchange resins (cholestyramine) or large amounts of calcium or magnesium chloride can result in increased absorp-tion of chloride and loss of bicarbonate ions. These nonabsorbable resins bind bicarbonate ions, whereas calcium and magnesium combine with bicarbonate to form insoluble salts within the intestines.
B. Increased Renal Loss of HCO3−
Renal wasting of HCO3− can occur as a result of fail-ure to reabsorb filtered HCO3− or to secrete adequate amounts of H+ in the form of titratable acid or ammo-nium ion. These defects are encountered in patients taking carbonic anhydrase inhibitors, such as acet-azolamide, and in those with renal tubular acidosis.
Renal tubular acidosis comprises a group ofnonazotemic defects of H+ secretion by the renaltubules, resulting in a urinary pH that is too high for the systemic acidemia. These defects may be a result of a primary renal defect or may be secondary to a systemic disorder. The site of the H+-secreting defect may be in the distal (type 1) or proximal (type 2) renal tubule. Hyporeninemic hypoaldosteronism is commonly referred to as type 4 renal tubular aci-dosis. With distal renal tubular acidosis, the defect occurs at a site after most of the filtered HCO3− has been reclaimed. As a result, there is a failure to acidify the urine, so that net acid excretion is less than the daily net acid production. This disorder is frequently associated with hypokalemia, deminer-alization of bone, nephrolithiasis, and nephrocalci-nosis. Alkali (NaHCO 3) therapy (1–3 mEq/kg/d) is usually sufficient to reverse those side effects. With the less common proximal renal tubular acidosis, defective H + secretion in the proximal tubule results in massive wasting of HCO3−. Concomitant defects in tubular reabsorption of other substances, such as glucose, amino acids, or phosphates, are com-mon. The hyperchloremic acidosis results in volume depletion and hypokalemia. Treatment involves giv-ing alkali (as much as 10–25 mEq/kg per day) and potassium supplements.
C. Other Causes of Hyperchloremic Acidosis
A dilutional hyperchloremic acidosis can occur when extracellular volume is rapidly expanded with a bicarbonate-free fluid, such as normal saline. This is a reason to prefer balanced salt solutions over 0.9% saline for fluid resuscitation. The plasma HCO3− decreases in proportion to the amount of fluid infused as extracellular HCO 3− is diluted. Amino acid infusions (parenteral hyperalimentation) con-tain organic cations in excess of organic anions and can produce hyperchloremic metabolic acidosis because chloride is commonly used as the anion for the cationic amino acids. Lastly, the administration of excessive quantities of chloride-containing acids, such as ammonium chloride or arginine hydrochlo-ride (usually given to treat a metabolic alkalosis), can cause hyperchloremic metabolic acidosis.
Treatment of Metabolic Acidosis
Several general measures can be undertaken to con-trol the severity of acidemia until the underlying processes are corrected. Any respiratory component of the acidemia should be corrected. Respiration should be controlled, if necessary; a Paco2 in the low 30s may be desirable to partially return pH to nor-mal. If arterial blood pH remains below 7.20, alkali therapy, usually in the form of NaHCO3(usually a 7.5% solution), may be necessary. Paco2 may tran-siently rise as HCO3− is consumed by acids (empha-sizing the need to control ventilation in severe acidemia). The amount of NaHCO3 given is decided empirically as a fixed dose (1 mEq/kg) or is derived from the base excess and the calculated bicarbon-ate space . In either case, serial blood gas measurements are mandatory to avoid complica-tions (eg, overshoot alkalosis and sodium overload) and to guide further therapy. Raising arterial pH to >7.25 is usually sufficient to overcome the adverse physiological effects of the acidemia. Profound or refractory acidemia may require acute hemodialysis with a bicarbonate dialysate.
The routine use of large amounts of NaHCO3 in treating cardiac arrest and low flow states is not recommended. Paradoxical intracellular acidosis may occur, particularly when CO2 elimination is impaired, because the CO2 formed readily enters cells, but the bicarbonate ion does not. Alternate buf-fers that do not produce CO2, such as CarbicarbTM or tromethamine (THAM), may be theoretically preferable, but are unproven clinically.
Specific therapy for diabetic ketoacidosis includes replacement of the existing fluid deficit (as a result of a hyperglycemic osmotic diuresis) first, as well as insulin, potassium, phosphate, and magnesium. The treatment of lactic acidosis should be directed first at restoring adequate oxygenation and tissue perfusion. Alkalinization of the urine with NaHCO 3 to a pH greater than 7.0 increases elimination of salicylate following salicylate poi-soning. Treatment options for ethanol or ethylene glycol intoxication include ethanol infusion or fomepizole administration, which competitively inhibit alcohol dehydrogenase and hemodialysis or hemofiltration.
Bicarbonate Space
The bicarbonate space is defined as the volume to which HCO3− will distribute when it is given intrave-nously. Although this theoretically should equal the extracellular fluid space (approximately 25% of body weight), in reality, it ranges anywhere between 25% and 60% of body weight, depending on the sever-ity and duration of the acidosis. This variation is at least partly related to the amount of intracellular and bone buffering that has taken place.
Example: Calculate the amount of NaHCO3necessary to correct a base deficit (BD) of –10 mEq/L for a 70-kg man with an estimated HCO3− space of 30%:
NaHCO3= BD × 30% × body weight in L
NaHCO3= −10 mEq/L × 30% × 70 L = 210 mEq
In practice, only 50% of the calculated dose (105 mEq) is usually given, after which another blood gas is measured.
Related Topics