Chapter: Organic Chemistry: Alkenes and alkynes
Properties of alkenes and alkynes
PROPERTIES OF ALKENES AND ALKYNES
Key Notes
Structure
Alkenes
are planar with bond angles of 120 . The carbon atoms of the C=C bond are sp2 hybridized and the double
bond is made up of one σ bond and one π bond. Alkynes are linear with the triple bond
carbons being sp hybridized. The
triple bond is made up of one σ bond and two π bonds.
C-C Bond
The C=C
bond is stronger and shorter than a C–C single bond. However, the two bonds
making up the C=C bond are not of equal strength. The π bond is weaker than the σ bond. Bond rotation round a C=C
bond is not possible and isomers are possible depending on the substituents
present. The more substituents which are present on an alkene, the more stable
the alkene is.
C=C Bond
An
alkyne triple bond is stronger than a C–C single bond or a C=C double bond. The
two π bonds
present in the triple bond are weaker and more reac-tive than the σ bond.
Properties
Alkenes
and alkynes are nonpolar compounds which dissolve in nonpolar sol-vents and are
very poorly soluble in water. They have low boiling points since only weak van
der Waals interactions are possible between the molecules.
Nucleophilicity
Alkenes
and alkynes act as nucleophiles and react with electrophiles by a reaction
known as electrophilic addition. The nucleophilic centers are the multiple
bonds which are areas of high electron density.
Spectroscopic analysis of alkenes
Alkenes
show characteristic C=C stretching absorptions in their IR spectra. Absorptions
due to C-H stretching and bending may also be identifiable. The latter can give
an indication of the substitution pattern. The nmr signals for alkene protons
and carbons occur in characteristic regions of the rele-vant nmr spectra.
Chemical shifts, coupling patterns and coupling con-stants can be used to
identify the stereochemistry of the alkene.
Spectroscopic analysis of alkynes
Unsymmetrical
alkynes show a characteristic triple bond stretching absorp-tion in their IR
spectra. This signal is absent for symmetrical alkynes. Terminal alkynes show a
characteristic C–H stretching absorption in their IR spectra as well as a
signal at low chemical shift in the 1H nmr. Alkyne carbons appear in
a characteristic region of the 13C nmr spectrum.
Structure
The alkene functional group (R2CCR2)
is planar in shape with bond angles of 120°. The two carbon atoms involved in the double
bond are both sp2
hybridized.
Each carbon has three sp2 hybridized orbitals which are used for σ bonds while the p orbital
is used for aπbond. Thus, the double bond is made up of oneσbond and one π bond (Fig.
1).
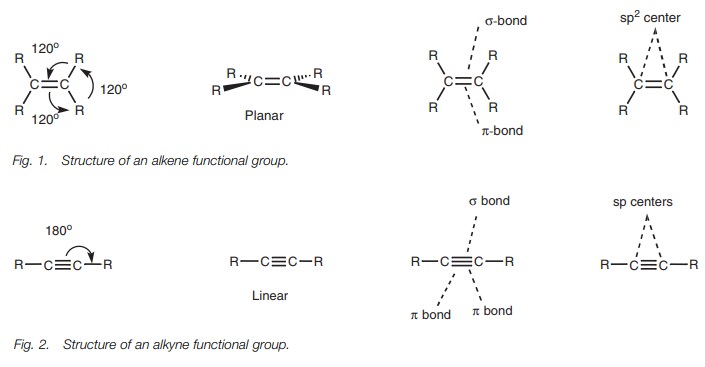
The alkyne functional group consists of a carbon
carbon triple bond and is linear in shape with bond angles of 180°(Fig. 2).
The two carbon atoms involved in the triple bond are both sp hybridized, such that each carbon atom has two sp hybridized orbitals and two p orbitals. The sphybridized orbitals are used for two σ bonds while the p orbitals are used for two π bonds. Thus, the triple bond is made up of one σ bond and two π bonds.
C = C Bond
The C = C bond is stronger (152 kcal mol-1)
and shorter (1.33 Å) than a C–C single bond (88 kcal mol -1 and 1.54 Å respectively). A C=C bond contains
one σ bond and one π bond, with the π bond being weaker than the σ bond. This
is important with respect to the reactivity of alkenes.
Bond rotation is not possible for a C = C
double bond since this would require the π bond to be broken. Therefore,
isomers of alkenes are possible depending on the relative position of the
substituents. These can be defined as cis or trans, but are more properly
defined as (Z) or (E).
Alkenes are defined as mono-, di-, tri-, or
tetrasubstituted depending on the number of substituents which are present. The
more substituents which are pre- sent, the more stable the alkene.
C = C Bond
The bond length of a carbon carbon triple bond
is 1.20 Å and the bond strength is 200 kcal mol
1. The π bonds are weaker than the σ bond. The presence of the π bonds
explains why alkynes are more reactive than alkanes.
Properties
Alkenes and alkynes have physical properties similar to alkanes. They arerelatively nonpolar, dissolve in nonpolar solvents and are not very soluble in water. Only weak van der Waals interactions are possible between unsaturated molecules such as alkene and alkynes, and so these structures have low boiling points compared to other functional groups.
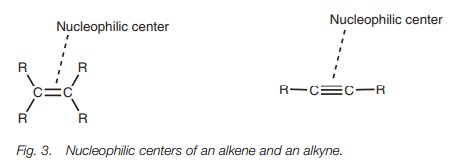
Nucleophilicity
Alkenes and alkynes are nucleophilic and
commonly react with electrophiles in a reaction known as electrophilic
addition. The nucleophilic center of the alkene or alkyne is the double bond or
triple bond (Fig. 3). These are areas of high electron density due to the
bonding electrons. The specific electrons which are used toform bonds to
attacking electrophiles are those involved in π bonding.
Spectroscopic analysis of alkenes
The IR spectrum of an alkene shows a
characteristic stretching absorption for the C=C bond
at 1680–1620 cm−1. This
is slightly higher
than the corresponding absorptions for aromatic rings.
However, conjugation with aromatic rings, carbonyl groups or other double bonds
can lower the absorption frequencyas far as 1590 cm−1. Some carbonyl
absorptions can occur in the same region butare generally stronger if they are
present.
The
C–H stretching absorption
of an alkene
proton occurs in the region 3095–3010 cm−1 but is
generally weak and could be difficult to spot. It can also be confused with the
C–H stretching absorptions for an aromatic ring. Alkene C–H out of plane
bending absorptions may sometimes be visible in the fingerprint region and can
indicate the substitution pattern. For example, an absorption in the region
730–675 cm−1 is indicative of
cis alkenes, while an absorption in the region 990–960 cm−1 is indicative of trans alkenes.
Monosubstituted alkenes have two absorptions at 910 and 990 cm−1.
The nmr signals for the protons and carbons of
an alkene group appear at char- acteristic positions in their respective
spectra (typically 4.5–6.5 ppm for 1H spectra and 80–145 ppm for 13C
spectra). The chemical shifts of each signal can indicate the stereochemistry
of the alkene, and nmr tables are available to work out the theoretical shifts
based on the substituents present and their relative positions. If coupling is
observed between the alkene protons in the proton spectrum, the cou- pling
pattern and the coupling constants can indicate the stereochemistry. For
example, the coupling constants for protons which are trans or cis with respect
to each other are typically 14–16 Hz and 6–8 Hz respectively. The coupling
constants for geminal protons (i.e. protons on the same alkene carbon) are
small (typically 1–2 Hz).
In the uv spectrum of ethene, the λmax for the
π−π* transition is 171 nm. For con- jugated systems the wavelength of this
transition increases and can indicate the amount of conjugation as well as the
substitution pattern.
Spectroscopic analysis of alkynes
The IR spectrum of an alkyne shows a
characteristic triple bond absorption in the region 2260–2100 cm−1.
This can often be weak and if the alkyne is symmetrical no absorption will be
seen at all. Although the absorption may be weak, it can usually be
identified since the
relevant region is
normally devoid of
peaks.
Terminal alkynes show an absorption at 3300 cm−1 due to C–H stretching. This is sharper than
absorptions due to O–H or N–H stretching.
The proton of a terminal alkyne typically gives
a signal at 1.8–3.1 ppm in the 1H nmr, while the carbons of any
alkyne appear in the region 70–95 ppm in the 13C nmr.
Related Topics