Chapter: Clinical Anesthesiology: Clinical Pharmacology: Pharmacological Principles
Pharmacokinetics: Distribution
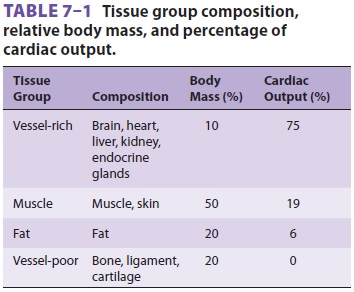
Distribution
Once absorbed, a drug is distributed by
the blood-stream throughout the body. Highly perfused organs (the so-called
vessel-rich group) receive a dispro-portionate fraction of the cardiac output (
Table 7–1).
Therefore, these tissues receive a disproportionate amount of drug in the first
minutes following drug administration. These tissues approach equilibration
with the plasma concentration more quickly than less well perfused tissues due
to the differences in
blood flow. However, less well perfused
tissues such as fat and skin may have enormous capacity to absorb lipophilic
drugs, resulting in a large reservoir of drug following long infusions.
Drug molecules obey the law of mass
action. When the plasma concentration exceeds theconcentration in tissue, the
drug moves from the plasma into tissue. When the plasma concentra-tion is less
than the concentration in tissue, the drug moves from the tissue back to
plasma.
Distribution is a major determinant of
end-organ drug concentration. The rate of rise in drug concentration in an
organ is determined by that organ’s perfusion and the relative drug solubility
in the organ compared with blood. The equilib-rium concentration in an organ
relative to blood depends only on the relative solubility of the drug in the
organ relative to blood, unless the organ is capable of metabolizing the drug.
Molecules in blood are either free or
bound to plasma proteins and lipids. The free concentration equilibrates
between organs and tissues. However, the equilibration between bound and
unbound mol-ecules is instantaneous. As unbound molecules of drug diffuse into
tissue, they are instantly replaced by previously bound molecules. Plasma
protein bind-ing does not affect the rate of transfer directly, but it does
affect relative solubility of the drug in blood and tissue. If the drug is
highly bound in tissues, and unbound in plasma, then the relative solubility
favors drug transfer into tissue. Put another way, a drug that is highly bound
in tissue, but not in blood, will have a very large free drug concentration
gradi-ent driving drug into the tissue. Conversely, if the drug is highly bound
in plasma and has few binding sites in the tissue, then transfer of a small
amount of drug may be enough to bring the free drug concen-tration into
equilibrium between blood and tissue. Thus, high levels of binding in blood
relative to tis-sues increase the rate of onset of drug effect, because fewer
molecules need to transfer into the tissue to produce an effective free drug
concentration.
Albumin binds many acidic drugs (eg,
barbi-turates), whereas α1-acid glycoprotein (AAG) binds basic drugs (local
anesthetics). If the concentrations of these proteins are diminished or
(typically less important) if the protein-binding sites are occupied by other
drugs, then the relative solubility of the drugs in blood is decreased,
increasing tissue uptake. Kidney disease, liver disease, chronic congestive
heart failure, and malignancies decrease albumin produc-tion. Trauma (including
surgery), infection, myocar-dial infarction, and chronic pain increase AAG
levels. Pregnancy is associated with reduced AAG concen-trations. Note that
these changes will have very little effect on propofol, which is administered
with its own binding molecules (the lipid in the emulsion).
Lipophilic molecules can readily
transfer between the blood and organs. Charged molecules are able to pass in
small quantities into most organs. However, the blood–brain barrier is a
special case. Permeation of the central nervous system by ionized drugs is
limited by pericapillary glial cells and endothelial cell tight junctions. Most
drugs that readily cross the blood–brain barrier (eg, lipophilic drugs like
hypnotics and opioids) are avidly taken up in body fat.
The time course of distribution of drugs
into peripheral tissues is complex and can only be assessed with computer
models. Following intra-venous bolus administration, rapid distribution of drug
from the plasma into peripheral tissues accounts for the profound decrease in
plasma con-centration observed in the first few minutes. For each tissue, there
is a point in time at which the apparent concentration in the tissue is the
same as the concentration in the plasma. The redistribu-tion phase (for each
tissue) follows this moment of equilibration. During redistribution, drug
returns from peripheral tissues back into the plasma. This return of drug back
to the plasma slows the rate of decline in plasma drug concentration.
Distribution generally contributes to
rapid emergence by removing drug from the plasma for many minutes following
administration of a bolus infusion. Following prolonged infusions, redistri-bution generally delays
emergence as drug returns from tissue reservoirs to the plasma for many hours.
The complex process of drug distribution
into and out of tissues is one reason that half-lives are clinically useless.
The offset of a drug’s clinical actions are best predicted by computer models
using the context-sensitive half-time or decrement times. The context-sensitive half-time is the time
required for a 50% decrease in plasma drug concentration to occur following a
pseudo steady-state infusion (in other words, an infusion that has continued
long enough to yield nearly steady-state concentrations). Here the “context” is
the duration of the infusion. The context-sensitive
decrement time is a more gen-eralized concept referring to any clinically
relevant decreased concentration in any tissue, particularly the brain or
effect site.
The volume of distribution, Vd,
is the appar-ent volume into which a
drug has “distributed” (ie,mixed). This volume is calculated by dividing a
bolus dose of drug by the plasma concentration at time 0. In practice, the
concentration used to define the Vd is often obtained by extrapolating subsequent
concentrations back to “0 time” when the drug was injected, as follows:

The concept of a single Vd
does not apply to any intravenous drugs used in anesthesia. All intra-venous
anesthetic drugs are better modeled with at least two compartments: a central
compartment and a peripheral compartment. The behavior of many of these drugs
is best described using three compartments: a central compartment, a rapidly
equilibrating peripheral compartment, and a slowly equilibrating peripheral
compartment. The central compartment may be thought of as including the blood
and any ultra-rapidly equilibrating tissues such as the lungs. The peripheral
compartment is composed of the other body tissues. For drugs with two
peripheral compartments, the rapidly equilibrat-ing compartment comprises the
organs and muscles, while the slowly equilibrating compartment roughly
represents distribution of the drug into fat and skin. These compartments are
designated V1 (central), V2(rapid distribution), and V3(slow distribution).The volume of
distribution at steady state, Vdss is the algebraic sum of these compartment volumes. V1
is calculated by the above equation showing the rela-tionship between volume,
dose, and concentration. The other volumes are calculated through
pharma-cokinetic modeling.
A small Vdss implies that the drug has high aqueous
solubility and will remain largely within the intravascular space. For example,
the Vdss
of pancuronium is about 15 L in a 70-kg person, indi-cating that pancuronium is
mostly present in body water, with little distribution into fat. However, the
typical anesthetic drug is lipophilic, resulting in a Vdssthat exceeds total body water
(approximately 40 L). For example, the Vdss for fentanyl is about 350 L in
adults, and the Vdss for propofol may exceed 5000 L. Vdss does not represent a
real volume but rather reflects the volume into which the drug would need to
distribute to account for the observed plasma concentration given the dose that
was administered.
Related Topics