Chapter: Clinical Anesthesiology: Clinical Pharmacology: Pharmacological Principles
Halothane - Clinical Pharmacology of Inhalation Anesthetics
HALOTHANE
Physical Properties
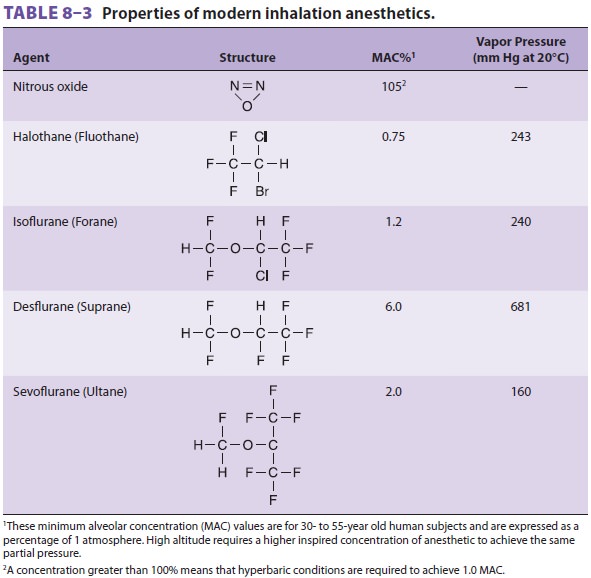
Halothane is a halogenated alkane (see
Table 8–3). The carbon–fluoride bonds are responsible for its nonflammable and
nonexplosive nature. Thymol preservative and amber-colored bottles retard
spon-taneous oxidative decomposition. It is rarely used in the United States.
Effects on Organ Systems
A. Cardiovascular
A dose-dependent reduction of arterial
blood pressure is due to direct myocardial depression; 2.0 MAC of halothane in
patients not undergoing surgery results in a 50% decrease in blood pres-sure
and cardiac output. Cardiac depression— from interference with sodium–calcium
exchange and intracellular calcium utilization—causes an increase in right
atrial pressure. Although halothane is a coronary artery vasodilator, coronary
blood flow decreases, due to the drop in systemic arterial pressure. Adequate
myocardial perfusion is usually maintained, as oxygen demand also drops.
Normally, hypotension inhibits baroreceptors in the aortic arch and carotid
bifurcation, causing a decrease in vagal stimulation and a compensatory rise in
heart rate. Halothane blunts this reflex. Slowing of sinoatrial node conduction
may result in a junctional rhythm or bradycardia. In infants, halothane
decreases car-diac output by a combination of decreased heart rate and
depressed myocardial contractility. Halothane sensitizes the heart to the
arrhythmogenic effects of epinephrine, so that doses of epinephrine above 1.5
mcg/kg should be avoided. Although organ blood flow is redistributed, systemic
vascular resistance is unchanged.
B. Respiratory
Halothane
typically causes rapid, shallow breath-ing. The increased respiratory rate is
not enough to counter the decreased tidal volume, so alveolar ventilation
drops, and resting Paco 2 is elevated. Apneic threshold, the highest Paco2at which apatient
remains apneic, also rises because the dif-ference between it and resting Paco2
is not altered by general anesthesia. Similarly, halothane limits the increase
in minute ventilation that normally accom-panies a rise in Paco2.
Halothane’s ventilatory effects are probably due to central (medullary
depression) and peripheral (intercostal muscle dysfunction) mechanisms. These
changes are exaggerated by preexisting lung disease and attenuated by surgical
stimulation. The increase in Paco2 and the decrease in intrathoracic
pressure that accompany spontane-ous ventilation with halothane partially
reverse the depression in cardiac output, arterial blood pres-sure, and heart
rate described above. Hypoxic drive is severely depressed by even low
concentrations of halothane (0.1 MAC).
Halothane
is considered a potent bronchodila-tor, as it often reverses asthma-induced
broncho-spasm. This action is not inhibited by β-adrenergic
blocking agents. Halothane attenuates airway reflexes and relaxes bronchial
smooth muscle by inhibiting intracellular calcium mobilization. Halothane also
depresses clearance of mucus from the respiratory tract (mucociliary function),
promoting postopera-tive hypoxia and atelectasis.
C. Cerebral
By dilating cerebral vessels, halothane
lowers cerebral vascular resistance and increases CBF. Autoregulation, the maintenance of constant CBFduring changes in
arterial blood pressure, is blunted. Concomitant rises in intracranial pressure
can be prevented by establishing hyperventilation priorto administration of halothane. Cerebral activity isdecreased,
leading to electroencephalographic slow-ing and modest reductions in metabolic
oxygen requirements.
D. Neuromuscular
Halothane relaxes skeletal muscle and
potentiates nondepolarizing neuromuscular-blocking agents (NMBA). Like the
other potent volatile anesthetics, it is a triggering agent of malignant
hyperthermia.
E. Renal
Halothane reduces renal blood flow,
glomerular fil-tration rate, and urinary output. Part of this decrease can be
explained by a fall in arterial blood pressure and cardiac output. Because the
reduction in renal blood flow is greater than the reduction in glomeru-lar
filtration rate, the filtration fraction is increased. Preoperative hydration
limits these changes.
F. Hepatic
Halothane causes hepatic blood flow to
decrease in proportion to the depression of cardiac output. Hepatic artery
vasospasm has been reported during halothane anesthesia. The metabolism and
clearance of some drugs (eg, fentanyl, phenytoin, verapamil) seem to be
impaired by halothane. Other evidence of hepatic cellular dysfunction includes
sulfobro-mophthalein (BSP) dye retention and minor liver transaminase
elevations.
Biotransformation & Toxicity
Halothane is oxidized in the liver by a
particular iso-zyme of CYP (2EI) to its principal metabolite, tri-fluoroacetic
acid. This metabolism can be inhibited by pretreatment with disulfiram.
Bromide, another oxidative metabolite, has been incriminated in (but is an improbable
cause of) postanesthetic changes in mental status. In the absence of oxygen,
reductive metabolism may result in a small amount of hepa-totoxic end products
that covalently bind to tissue macromolecules. This is more apt to occur
following enzyme induction by phenobarbital. Elevated fluo-ride levels signal
significant anaerobic metabolism.
Postoperative hepatic dysfunction has
several causes: viral hepatitis, impaired hepatic perfusion, preexisting liver
disease, hepatocyte hypoxia, sepsis, hemolysis, benign postoperative
intrahepatic cholestasis, and drug-induced hepatitis. “Halothanehepatitis” is extremely rare (1 per 35,000 cases).
Patients exposed to multiple halothane
anesthetics at short intervals, middle-aged obese women, and persons with a
familial predisposition to halothane toxicity or a personal history of toxicity
are considered to be at increased risk. Signs are mostly related to hepatic
injury, such as increased serum alanine and aspartate transferase, elevated
bilirubin (leading to jaundice), and encephalopathy.
The hepatic lesion seen in
humans—centrilobular necrosis—also occurs in rats pretreated with an enzyme
inducer (phenobarbital) and exposed to halo-thane under hypoxic conditions (Fio2< 14%). This halothane
hypoxic model implies hepatic damage fromreductive metabolites or hypoxia.
More likely evidence points to an immune
mechanism. For instance, some signs of the dis-ease indicate an allergic
reaction (eg, eosinophilia, rash, fever) and do not appear until a few days
after exposure. Furthermore, an antibody that binds to hepatocytes previously
exposed to halothane has been isolated from patients with halothane-induced
hepatic dysfunction. This antibody response may involve liver microsomal
proteins that have been modified by trifluoroacetic acid as the triggering
antigens (trifluoroacetylated liver proteins such as microsomal
carboxylesterase). As with halothane, other inhalational agents that undergo
oxidative metabolism can likewise lead to hepatitis. However, newer agents
undergo little to no metabolism, and therefore do not form trifluroacetic acid
protein adducts or produce the immune response leading to hepatitis.
Contraindications
It is prudent to withhold halothane from
patients with unexplained liver dysfunction following previ-ous anesthetic
exposure. Halothane, like all inhalational anesthetics, should be used with
care in patients with intracranial mass lesions because of the possibility of
intracranial hypertension secondary to increased cerebral blood volume and
blood flow.Hypovolemic patients and some patients with severe reductions in
left ventricular function may not tolerate halothane’s negative inotropic
effects. Sensitization of the heart to catecholamines lim-its the usefulness of
halothane when exogenous epinephrine is administered or in patients with
pheochromocytoma.
Drug Interactions
The myocardial depression seen with
halothane is exacerbated by β-adrenergic-blocking agents and calcium
channel-blocking agents. Tricyclic antide-pressants and monoamine oxidase
inhibitors have been associated with f uctuationsl in blood pres-sure and
arrhythmias, although neither represents an absolute contraindication. The
combination of halothane and aminophylline has resulted in serious ventricular
arrhythmias.
Related Topics