Chapter: Clinical Anesthesiology: Clinical Pharmacology: Pharmacological Principles
Pharmacokinetics: Compartment Models
Compartment Models
Multicompartment models provide a
mathemati-cal framework that can be used to relate drug dose to changes in drug
concentrations over time. Conceptually, the compartments in these models are
tissues with a similar distribution time course. For example, the plasma and
lungs are components of the central compartment. The organs and muscles,
sometimes called the vessel-rich group, could be the second, or rapidly
equilibrating, compartment. Fat and skin have the capacity to bind large
quan-tities of lipophilic drug but are poorly perfused. These could represent
the third, or slowly equili-brating, compartment. This is an intuitive
definition of compartments, and it is important to recognize that the
compartments of a pharmacokinetic model are mathematical abstractions that
relate dose to observed concentration. A one-to-one relationship does not exist
between any compartment and any organ or tissue in the body.
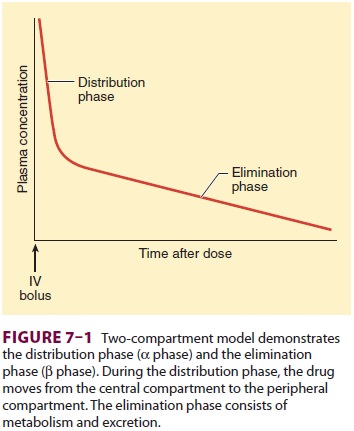
Many drugs used in anesthesia are well
described by a two-compartment model. This is generally the case if the studies
used to characterize the pharmacokinetics do not include rapid arterial
sampling over the first few minutes (Figure 7–1). Without rapid arterial sampling
the ultra-rapid ini-tial drop in plasma concentration immediately after a bolus
injection is missed, and the central compart-ment volume is blended into the
rapidly equilibrat-ing compartment. When rapid arterial sampling is used in
pharmacokinetic experiments, the results are generally a three-compartment model.
In these cases the number of identifiable compartments is a function of the
experimental design and not a char-acteristic of the drug.
In compartmental models the
instantaneous concentration at the time of a bolus injection is assumed to be
the amount of the bolus divided by the central compartment volume. This is not
correct. If the bolus is given over a
few seconds, the instantaneous concentration is 0, because the drug is all in
the vein, still flowing to the heart. It takes only a minute or two for the
drug to mix in the cen-tral compartment volume. This misspecification is common
to conventional pharmacokinetic mod-els. More physiologically based models,
sometimes called front-end kinetic models,
can characterize the initial delay in concentration. These models are useful
only if the concentrations over the first few minutes are clinically important.
After the first few minutes, front-end models resemble conventional
compartmental models.
In the first few minutes following
initial bolus administration of a drug, the concentration drops very rapidly as
the drug quickly diffuses into periph-eral compartments. The decline is
typically an order of magnitude over 10 minutes. For drugs with very rapid hepatic
clearance (eg, propofol) or those that are metabolized in the blood (eg,
remifentanil), metabolism contributes significantly to the rapid ini-tial drop
in concentration. Following this very rapid drop there is a period of slower
decrease in plasma concentration. During this period, the rapidly equilibrating
compartment is no longer removing drug from the plasma. Instead, drug returns
to the plasma from the rapidly equilibrating compartment. The reversed role of
the rapidly equilibrating tissues from extracting drug to returning drug
accounts for the slower rate of decline in plasma concentration in this
intermediate phase. Eventually there is an even slower rate of decrease in
plasma concentration, which is log-linear until the drug is completely elim-inated
from the body. This terminal log-linear phase occurs after the slowly
equilibrating compartment shifts from net removal of drug from the plasma to
net return of drug to the plasma. During this ter-minal phase the organ of
elimination (typically the liver) is exposed to the body’s entire body drug
load, which accounts for the very slow rate of decrease in plasma drug
concentration during this final phase.
The mathematical models used to describe
a drug with two or three compartments are, respectively:
Cp(t) = Ae−αt+ Be−βt
and
Cp(t) = Ae−αt+ Be−βt+ Ce−γt
where Cp(t) equals plasma
concentration at time t, and α, β, and γ are the
exponents that character-ize the very rapid (ie, very steep), intermediate, and
slow (ie, log-linear) portions of the plasma concen-tration over time,
respectively. Drugs described by two-compartment and three-compartment models
will have two or three half-lives. Each half-life is cal-culated as the natural
log of 2 (0.693), divided by the exponent. The coefficients A, B,
and C represent the contribution of
each of the exponents to the overall decrease in concentration over time.
The two-compartment model is described
by a curve with two exponents and two coefficients, whereas the
three-compartment model is described by a curve with three exponents and three
coeffi-cients. The mathematical relationships among com-partments, clearances,
coefficients, and exponents are complex. Every coefficient and every exponent
is a function of every volume and every clearance.
Elimination half-life is the time
required for the drug concentration to fall by 50%. For drugsdescribed by
multicompartment pharmacokinetics (eg, all drugs used in anesthesia), there are
multiple elimination half-lives, in other words the elimination half-time is
context dependent. The offset of a drug’s effect cannot be predicted from
half-lives. Moreover, one cannot easily determine how rapidly a drug effect
will disappear simply by looking at coefficients, exponents, and half-lives.
For example, the terminal half-life of sufentanil is about 10 h, whereas that
of alfentanil is 2 h. This does not mean that recovery from alfentanil will be
faster, because clinical recovery from clinical dosing will be influ-enced by
all half-lives, not just the terminal one. Computer models readily demonstrate
that recovery from an infusion lasting several hours will be faster when the
drug administered is sufentanil than it will be when the infused drug is
alfentanil. The time required for a 50% decrease in concentration depends on
the duration or “context” of the infusion. The con-text-sensitive half-time,
discussed earlier, captures this concept and should be used instead of
half-lives to compare the pharmacokinetic properties of intra-venous drugs used
in anesthesia.
Related Topics