Chapter: Biology of Disease: Cancer
Molecular Biology of Cancer
MOLECULAR BIOLOGY OF CANCER
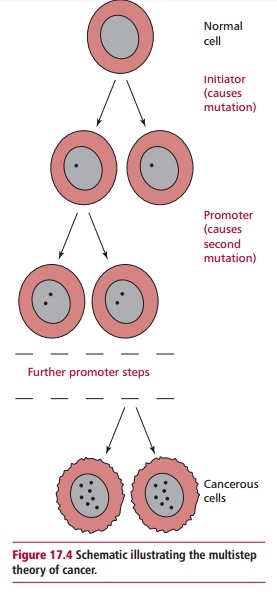
In 1914, Boveri (1862–1915) suggested that a malignant tumor arose from a cell that had acquired chromosomal abnormalities. In other words, cancer was caused by mutations in the DNA of cells. The multistep theory of cancer suggests that between five and seven successive mutations may have to occur before a cancer can develop (Figure 17.4). Each successive mutation gives the cell a selective growth advantage over the normal cells which surround it. For example, one mutation may confer the ability to grow at a faster rate than normal cells. The cell would then proliferate to form a tissue which is said to be hyperplastic. One of the daughter cells may then undergo another mutation, which allows uncontrolled division of the progeny, to form a mass of cells which have abnormal morphology. At this stage the tumor is known as a carcinoma in situ. Accumulation of further mutations within the clone may mean that the cells can detach from the tumor and invade surrounding tissue, that is, they metastasize.
In 2004, a census of published scientific literature showed that 291 genes, representing approximately 1% of the total number of human genes, are associated with cancers. For 90% of these genes, somatic mutations were detected in the cancer cells but not in normal tissue. Approximately 20% of ‘cancer genes’ show mutations in the germline DNA, which predispose an individual to cancer, while 10% have been associated with mutations in both germline and somatic DNA.
Genes that are associated with cancer include those that control the cell cycle, including cell division and differentiation or molecules involved in signal transduction and associated growth factors, as well as those that control the process of programmed cell death that results in apoptosis . Mutations in some of these genes may lead to increased proliferation and failures in apoptosis. Two major groups of genes linked to cancers are the oncogenes and the tumor suppressor genes.
ONCOGENES
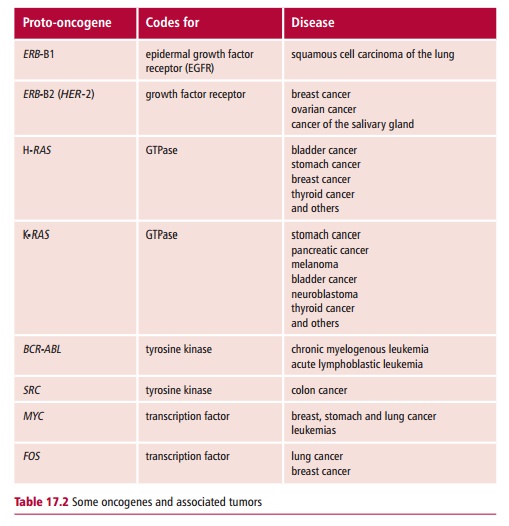
Oncogenes are mutated forms of normal genes, called proto-oncogenes, which stimulate the increased proliferation of abnormal cells by encoding factors including growth factors and receptors, as well as proteins involved in signal transduction . Examples include MYC, FOS and the RAS family of oncogenes. The mutation of a proto-oncogene to form an oncogene usually results in the production of a protein that has increased activity, or in the synthesis of greater than normal amounts of the protein, as, for example, when the gene is continually active. Such mutations are otherwise known as ‘gain-of-function’ mutations. Oncogenes were first discovered in certain viruses that cause cancer. Some viruses which cause cancer have a gene that is the equivalent of a cellular proto-oncogene. These genes are thought to have been derived initially from host DNA into which the viral DNA was integrated and to have undergone mutation during viral passage. When DNA derived from the virus becomes inserted into the host genome, the viral oncogene may provide the additional signal for cellular proliferation, or may override the normal cellular controls, resulting in the unregulated division of infected cells. The viral and cellular forms of the oncogene are usually distinguished by the prefix ‘v’ and ‘c’ respectively as in, for example, v-MYC and c-MYC.
Some examples of oncogenes and associated tumors are shown in Table 17.2.
TUMOR SUPPRESSOR GENES
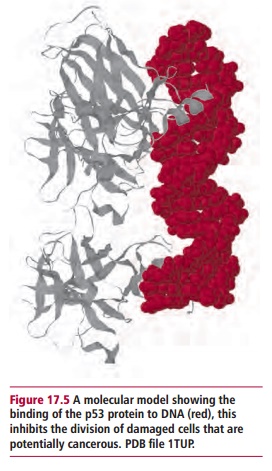
Tumor suppressor genes, in contrast to proto-oncogenes, encode proteins that inhibit the proliferation of cells that contain deleterious mutations. Mutations in the tumor suppressor genes themselves may then lead to a loss of this inhibition that is, they are ‘loss of function’ mutations. Some examples of tumor suppressor genes are the TP53gene, the retinoblastoma susceptibility gene, RB, and the Wilms’ tumor gene, WT1. The TP53 gene is found on the short arm of chromosome 17 and encodes a phosphoprotein, called tumor suppressor protein p53 (Figure 17.5) which has been called the ‘guardian of the genome’. The gene becomes activated in cells where DNA has become damaged, leading to the production of p53 protein. The protein can bind to DNA, blocking division of damaged cells and inducing apoptosis, thus preventing replication of potential tumors. Mutations in TP53 can lead to the production of a defective p53 which cannot recognize binding sites on DNA. Thus the replication of the cell is not inhibited, leading to a failure to remove damaged and potentially malignant cells.
Related Topics