Chapter: Biology of Disease: Cancer
Causes of Cancer
CAUSES OF CANCER
The mutations that lead to cancerous states are caused by or associated with a number of factors. These include mutations caused by errors in replicating DNA or failures in repairing damaged DNA, or they may be induced by a variety of environmental agents, including chemical carcinogens and ionizing radiation or by the action of some viruses. Mutations in cancer associated germ-line genes may also mean that offspring carrying the gene are more susceptible to developing cancer.
INHERITED CANCERS
The association between the inheritance of a mutated gene and the development of cancer genes may be direct or indirect. For a direct association, the offspring must inherit a mutated gene which confers increased susceptibility to a specific type of cancer. With an indirect association, the inherited gene is associated with defective mechanisms for the repair of DNA that, in turn, leads to a greater likelihood of cancer. An example of the latter occurs in people with the disease xeroderma pigmentosum in whom a failure to repair ultraviolet light-induced mutations in the DNA of skin cells leads to the development of multiple skin cancers.
Examples of cancer associated genes with a direct effect include the genes BRCA1 and BRCA2. Mutations in these genes, which are associated with anincreased susceptibility to cancer of the breast and ovaries, account for less than 2% of all breast cancers. However, patients with a defective BRCA gene have a much greater risk of developing breast cancer than those who do not. BRCA1 was mapped to chromosome 17q21 in 1990 and at least 100 mutationshave been identified in the gene. Women who inherit a mutated BRCA1 gene have a 60–83% chance of contracting breast cancer at some stage and a 20– 40% chance of developing ovarian cancer. This gene has now been sequenced and its product identified. It encodes a transcription factor (Figure 17.6) that regulates the expression of, amongst others, the tumor suppressor geneTP53. Thus the presence of a mutated gene encoding a defective transcription factor leads to a failure to eliminate damaged cells. Women carrying a mutated form ofBRCA1 may be offered a prophylactic bilateral mastectomy, that is the removal of the apparently healthy breasts to prevent the development of breast cancer.
The BRCA2 gene has been mapped to chromosome 13q12. Mutations in this gene confer a 40–60% chance of developing breast cancer and a 10–20% risk of ovarian cancer at some stage in their lives. A mutated BRCA2 gene also increases the risk for male breast cancer.
Another gene associated with breast cancer is CHK2, which encodes a protein kinase, called the checkpoint kinase. This enzyme is involved in the control of the cell cycle Inheriting the abnormal variant of CHK2 doubles the risk of a woman developing breast cancer, in that the variant form is present in 1.9% of women with breast cancer and 0.7% of healthy women.
CHROMOSOMES AND CANCER
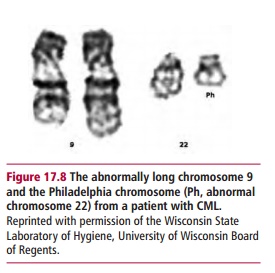
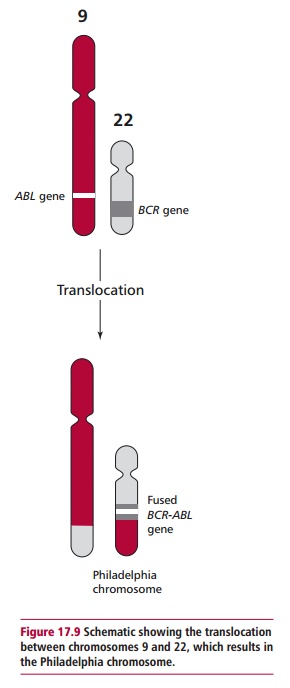
Chromosomal abnormalities are found in some cancers. For example, about 95% of patients with chronic myelogenous leukemia (CML) have a translocation involving chromosomes 9 and 22, that is, t(9:22). The translocation results in the production of a longer chromosome 9 and a shorter chromosome 22, commonly called the Philadelphia or Ph chromosome (Figure 17.8). The translocation results in the BCR gene on chromosome 22 becoming fused with part of the ABL gene on chromosome 9. The fused BCR-ABL encodes a tyrosine kinase that is continuously expressed, leading tocontinuous stimulation of proliferation and the development of CML (Figure17.9). The Philadelphia chromosome is also found in 25–30% of adults withacute lymphoblastic leukemia. In patients, the translocation originally occured in a single bone marrow cell. However, clonal expansion of the cell results in the blood becoming populated with cells bearing the Philadelphia chromosome. During active disease additional chromosomal abnormalities appear and these are indicative of a poor prognosis.
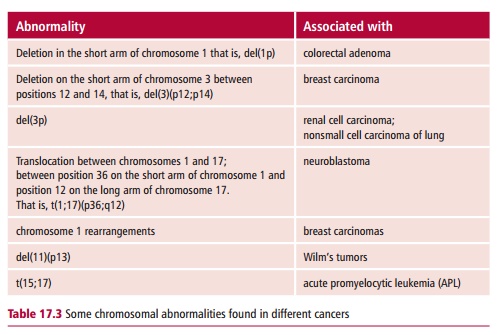
Other chromosome abnormalities have been associated with a wide range of cancers, including breast cancer, prostate cancer and neuroblastoma, but none to the same extent as that found in CML, where the presence of the Ph chromosome is diagnostic for the disease. Some other examples of chromosome abnormalities are shown inTable 17.3.
CHEMICAL CARCINOGENS
In 1775 Potts (1714–1788), an English surgeon, noted the high incidence of scrotal cancer amongst chimney sweeps in London and suggested that this may be related to the accumulation of soot in their clothes. As a result of legislation introduced to ensure that chimney sweeps were able to bathe and to change their clothing regularly, scrotal cancer was eliminated in this profession. In 1918, the Japanese scientists, Yamagiwa and Ichikawa showed that they could induce tumors experimentally by painting coal tar on to the skin of rabbits. This ability of certain compounds to induce tumors experimentally (Figure17.10) led to the identification of many carcinogenic chemicals, includingthose listed in Table 17.4.
The chemical induction of cancers is assumed to be a multistep process, probably involving mutations in several genes, possibly on different chromosomes. Carcinogenic compounds interact with DNA usually by one of a limited number of reactions. For example, alkylating agents such as dialkylnitrosamines and aflatoxin B1 lead to the addition of an alkyl group to electron-rich sites in DNA, as shown for aflatoxin B1 in Figure 12.4 (B). Aromatic amines and amides form highly electrophilic aryl nitrenium ions which also interact with DNA. Polycyclic aromatic hydrocarbons transfer an alkyl group to DNA.
At its simplest, the process of chemical carcinogenesis can be thought to occur in three phases namely: tumor initiation, promotion and progression. During tumor initiation, the carcinogen, or a metabolite of the carcinogen, produces a mutation in the DNA. The cell may repair the damage but misrepair may lead to heritable changes. A cell that has undergone initiation, however, is not yet cancerous because the cell has to ‘escape’ from normal growth control and become autonomous. Tumor promoters stimulate clonal proliferation of an initiated cell. An example of a promoter is tetradecanoyl phorbol acetate, a constituent of croton oil. Croton oil will promote the development of carcinomas in the skin of mice that have been treated with a single dose of benzo[a]pyrene. Tumor progression involves the additional changes that lead to malignancy and the ability to form metastases.
The targets for chemical carcinogens are the proto-oncogenes and the tumor suppressor genes described. Mutations in TP53 have been found in more than 500 types of human tumor. The mutations occur at vulnerable sites known as ‘hotspots’, though the position of the hotspot is not the same for all carcinogens. For example, the metabolite of benzo[a]pyrene preferentially forms adducts with guanine bases (Figure 12.4 (A)) at codons 157, 248 and 273 of TP53, which are the same mutational hotspots found in human lung cancers. This therefore supports the link between smoking and lung cancer that was established in the 1950s. Some chemical carcinogens may work by promoting the generation of reactive oxygen intermediates which themselves attack DNA. Thus health food shops promote the sale of supplements which are known to remove or ‘scavenge’ these free radicals. Alternatively some carcinogens may interfere more directly with the regulation of cell proliferation or receptor-mediated cell signaling processes.
Testing for potential carcinogens
The commonest test for the ability of chemicals to cause mutations in DNA is the Ames test which uses a mutant of Salmonella typhimurium that is unable to grow on growth media in the absence of the amino acid histidine (Figure 17.11). The test involves exposing the bacterium to the chemical in question, usually in the presence of an extract of mammalian liver to provide enzymes that may activate any procarcinogens present. Mutations caused by the potential carcinogen may result in the reversion of the mutant bacterium into one that can synthesize histidine and thus grow on the histidine deficient medium.
Mutagenicity can also be tested by determining the ability of the chemical in question to cause cytogenetic changes in the bone marrow of rodents. Traditional tests for carcinogenicity have ultimately relied on the use of laboratory animals, though this has obvious ethical implications. Some chemicals result in the induction of tumors in the majority of experimental animals after a single dose. The required time for a tumor to develop is known as the latency period and this may be shortened by administering several doses of the carcinogen.
DIET AND CANCER
It has been estimated by bodies such as the World Cancer Research Fund that between 30 and 40% of cancers could be prevented by eating a healthy diet, and by maintaining a healthy body weight, and participating in adequate physical activity. Conversely, prospective studies, in which researchers analyzed the diet and activity of a group of individuals, then monitored the frequency of cancer deaths in that group, have indicated that being overweight or obese contributes to 14% and 20% of deaths in men and women respectively. In addition, obesity has been strongly linked to a variety of cancers, including those of the GIT, liver, prostate, breast, uterus, cervix and ovary. There is also evidence to suggest a link between the consumption of foods with a high glycemic index and an increased incidence of cancer.
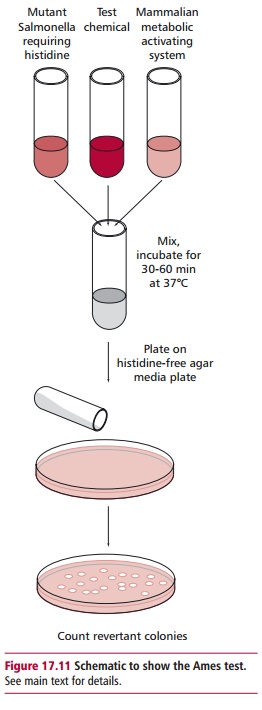
The consumption of low fiber, highly processed foods has a well-established association with the incidence of colorectal cancer though the link may be more complex than was originally thought. Indeed, an increased consumption of fiber-rich foods, such as fresh fruit and vegetables, has been correlated with a reduced incidence of several types of cancer, including those of the mouth, esophagus, lung and stomach in addition to those of the colon and rectum. Such associations have led to the recommendation by the UK Department of Health that individuals should consume at least five portions of fruit and vegetables a day. Cruciferous vegetables such as cabbage, cauliflower, broccoli and brussel sprouts contain sulfurophane (Figure 17.13), a chemical with anticancer properties. Increased consumption of such vegetables is inversely related to the incidence of breast and bladder cancers.
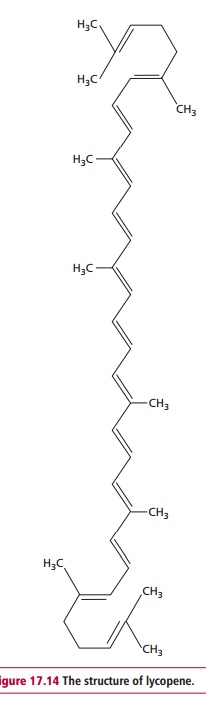
A number of nutritional interventions appear to offer some protection against cancer. These include decreasing the consumption of red meat and processed red meat, the intake of which is significantly linked to colorectal cancer. Increasing the consumption of W-3 fats such as α -linolenic acid, (ALA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) and decreasing that of W-6 fats such as linoleic acid is also recommended. Flax seeds, which are a good source both ofdietary fiber and ALA, have been shown to reduce the development of carcinogen-induced tumors and to reduce the rates of metastasis in animal models. Numerous studies have indicated that a sufficient vitamin D status offers protection against a variety of cancer types, and that improvements in its intake by, for example supplementation, could reduce the incidence of cancer and the associated mortality. Consumption of selenium, which is present in the active sites of several enzymes including the antioxidant enzyme glutathione peroxidase , has been associated with a decreased risk of prostate cancer in men. Other studies have suggested that antioxidants, such as those found in green tea, are protective against cancer, although this is controversial. In addition a high consumption of lycopene (Figure 17.14), a carotenoid found in tomatoes, has been shown in several studies to be associated with a decreased incidence of prostate cancer.
RADIATION AND CANCER
Ionizing radiation can promote the production of tumors in vivo, and the transformation of cultured cells in vitro from a normal to a malignant phenotype. Some of the evidence for radiation-induced carcinogenesis comes from studies of Japanese people irradiated during the atomic bomb explosions in Hiroshima (Figure 17.15) and Nagasaki, and from populations irradiated in nuclear accidents such as that which occurred in Chernobyl in 1986. Long-term studies of Japanese atom bomb survivors showed an increase in the incidence of leukemias in the first 5–10 years following exposure. The risk of solid tumors in these people was also increased significantly. Studies by Doll showed that infants previously subjected to X-irradiation in utero had an increased risk of developing leukemias and solid tumors.

The condition xeroderma pigmentosum also provides evidence for radiation-induced carcinogenesis. These patients suffer multiple skin cancers caused by the failure of their cells to repair ultraviolet light-induced damage to DNA. In addition, normal cells in culture may be transformed by irradiation into cells with a cancerous phenotype. Such studies have been used to analyze the nature of radiation-induced carcinogenesis.
DNA is also the target of ionizing radiation in radiation-induced carcinogenesis and the damage caused includes deletions, inversions and translocations Irradiation is also known to induce gene amplification and to increase chromosomal instability and these, in turn, increase the likelihood of mutations occurring. Ultimately, mutation events involving proto-oncogenes and/or tumor suppressor genes are the most likely causes of radiation-induced carcinogenesis.
With some exceptions, cells are more susceptible to radiation-induced damage when dividing and this fact has been utilized in the use of radiation to treat cancer.
VIRUSES AND CANCER
Viruses were first implicated in the development of some types of cancer when it was shown by Rous (1879–1970) in 1911 that leukemia in chickens was caused by a ‘filterable agent’. This virus, which causes sarcomas in chickens, is now called the Rous sarcoma virus and has been used extensively in research into oncogenic, or cancer-inducing, viruses.
Today, viruses are associated with between 10 and 20% of all human cancers. In the 1960s, Burkitt’s lymphoma, which is a tumor found in the jaws of children in certain regions of Africa, was found to be induced by a virus later identified as the Epstein-Barr virus (EBV; Figure 17.16) or human herpesvirus 4 (Table3.1) which also causes glandular fever, or infectious mononucleosis. TheEBV infects epithelial cells and B lymphocytes, causing their transformation. This virus is associated with a number of cancers, including nasopharyngeal carcinoma and Hodgkin’s lymphoma, in addition to Burkitt’s lymphoma.
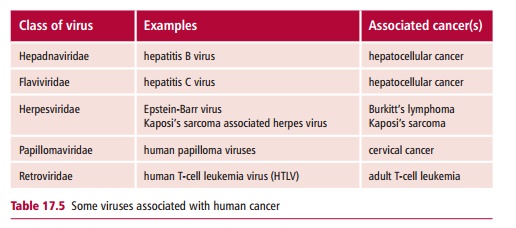
Cancer associated viruses belong to several groups, including the retroviruses (retroviridae), the papillomaviruses (papillomaviridae), the hepadnaviruses (hepadnaviridae), the flaviviridae and the herpesviridae (Table 17.5).
Retroviruses are RNA containing viruses that have reverse transcriptase activity. When they infect a cell, the reverse transcriptase transcribes their RNA into DNA which may become incorporated into the host genome. Not all retroviruses cause cancer but those which do are called ‘transforming retroviruses’. The human immunodeficiency virus (HIV) is a retrovirus that is also associated with cancer, although in this case the association is indirect. People with HIV who develop full-blown acquired immunodeficiency syndrome (AIDS;) have an increased incidence of tumors, such as lymphoma and Kaposi’s sarcoma, which are associated with EBV and Kaposi’s sarcoma virus (KSV). It is likely that the immunosuppression caused by HIV allows latent viruses such as EBV to escape immunological control. Kaposi’s sarcoma virus is also associated with nonHodgkin’s lymphoma and oral squamous cell carcinoma.

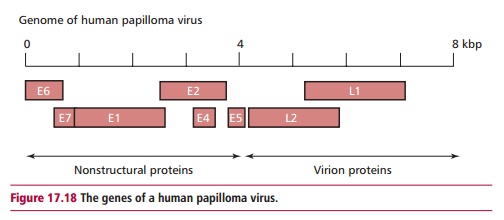
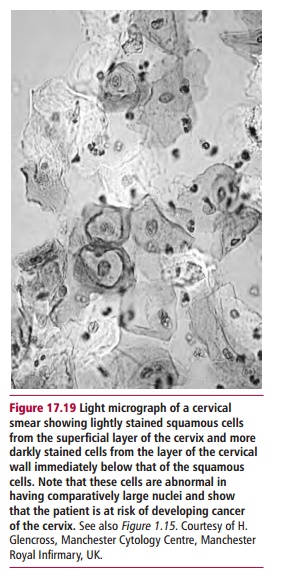
The most widely recognized association between viruses and cancer occurs with certain strains of human papilloma virus (HPV; Figure 17.17) that are linked to the development of cervical cancer. More than 70 papilloma viruses have been found in humans. The genomes of these viruses all have a similar structure and contain at least seven ‘early’ genes (E1–E7) and two ‘late’ genes, L1 and L2 (Figure 17.18). Some HPV subtypes invade epithelial cells of the skin, causing warts while others infect the genital tract and cause benign warts, with low risk of cancer. Others, such as HPV 16, 18, 31, 33 and 45, are associated with the development of cervical cancer in women and are regarded as ‘high risk’ for inducing cancer. DNA from at least one of these ‘high-risk’ types is detected in approximately 90% of human cervical cancers. The HPV E6 and E7 genes are
viral oncogenes and the proteins they encode inactivate certain regulators of cell division, such as the tumor suppressor protein p53. Cervical cancers develop from precursor ‘lesions’ that are graded high to low according to how much disruption of epithelial cell differentiation has occurred (Figure17.19). In cervical carcinomas, viral DNA becomes integrated into the hostgenome. However, many benign lesions also contain these strains of HPV, so the presence of the virus alone is insufficient to cause cancer. It seems that long-term infection with HPV predisposes the individual to cervical cancer and that other agents, for example the carcinogens in cigarette smoke, are required to allow the tumor to progress to full malignancy.
Related Topics