Chapter: Genetics and Molecular Biology: RNA Polymerase and RNA Initiation
Measuring the Activity of RNA Polymerase
Measuring the Activity of RNA Polymerase
Studies on protein synthesis in E. coli
in the 1960s revealed that a transient RNA copy of DNA is sent to ribosomes to
direct protein synthesis. Therefore cells had to contain an RNA polymerase that
was capable of synthesizing RNA from a DNA template. This property was sufficient
to permit enzymologists to devise assays for detection of the enzyme in crude
extracts of cells.
The original assay of RNA polymerase was merely a
measurement of the amount of RNA synthesized in vitro. The RNA synthesized was easily determined by measuring
the incorporation of a radioactive RNA pre-cursor, usually ATP, into a polymer
(Fig. 4.1). After synthesis, the radioactive polymer was separated from the
radioactive precursor nu-cleotides by precipitation of the polymer with acid,
and the radioactivity in the polymer was determined with a Geiger counter.
The precipitation procedure measures total
incorporated radioactiv-ity. It is adequate for the assay of RNA polymerase
activity and can be used to guide steps in the purification of the enzyme, but
it is indiscrimi-
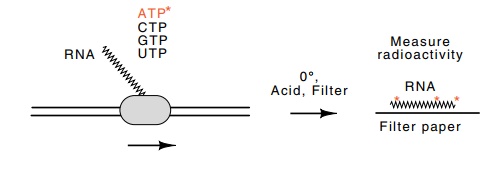
Figure
4.1 Assay of RNA polymerase by
incorporation of radioactive nucleo-tides into RNA in a reaction containing
buffer, NaCl, MgSO4, and triphosphates. After transcription, radioactive RNA and
radioactive nucleotides remain. These are separated by addition of
trichloroacetic acid and filtration. The filter paper then contains the RNA
whose radioactivity is quantitated in a Geiger counter or scintillation
counter.
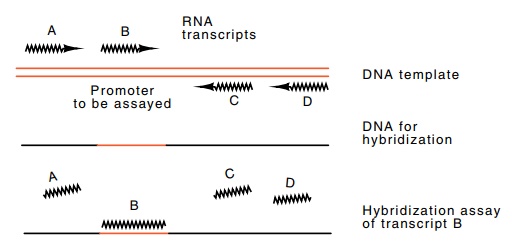
Figure
4.2 Assaying for specific
transcription by using a template and DNA forhybridization whose only sequences
in common are the region whose transcript is to be assayed.
nate. Only the total amount of RNA synthesized in
the reaction tube is quantitated. Since many of the convenient DNA templates
contain more than one promoter and since in
vitro transcription frequently initiates from random locations on the DNA
in addition to initiating from the promoters, a higher-resolution assay of
transcription is required to study specific promoters and the proteins that
control their activities.
Several basic methods are used to quantitate the
activities of specific promoters. One is to use DNA that contains several
promoters and specifically fish out and quantitate just the RNA of interest by
RNA-DNA hybridization (Fig. 4.2). Run-off transcription is another method that
is often used for examining the activity of promoters. This permits the
simultaneous assay of several promoters as well as nonspecific tran-scription.
Small pieces of DNA 200 to 2,000 base pairs long and contain-
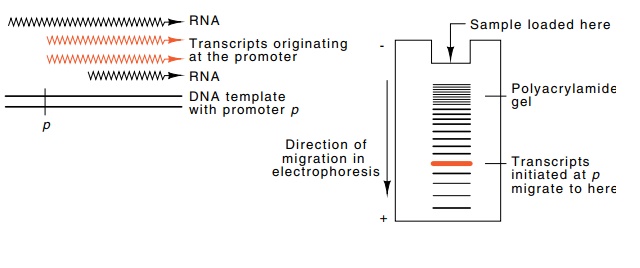
Figure
4.3 Assaying specific transcription
by electrophoresis on polyacry-lamide gels. The radioactive RNAs of different
sizes are synthesized using radioactive nucleoside triphosphates. Then the DNAs
are separated according to size by electrophoresis and their positions in the
gel found by autoradiogra-phy.
ing the promoter in question can be isolated.
Transcription initiating from the promoter on these templates begins at the
promoter and usually extends to the end of the DNA. This produces a small
transcript of a unique size. Transcription initiating from other sites on the
DNA generates other sizes of RNA transcripts. The resultant RNA molecules,
whose sizes vary from 10 to 1,000 nucleotides, may easily be separated from one
another by electrophoresis in polyacrylamide gels in the presence of high
concentrations of urea (Fig. 4.3). These denaturing agents reduce the formation
of transient hairpins in the RNA resulting from partial complementarity between
portions of the molecules. Therefore the RNA polymers migrate at velocities
dependent on their length and independent of their sequences. If α-32PO4
triphosphates have been used during the transcription, the locations of RNA
molecules in the gel can subsequently be determined by autoradiography.
The third
basic approach for examining the activity of a particular promoter became
possible through improvements in recombinant DNA technology. Small DNA
fragments several hundred nucleotides long can be isolated and used as
templates in transcription assays. Quantitation of the total RNA synthesized in
these reactions permits assay of a single promoter since the small DNA
templates usually contain only one promoter.
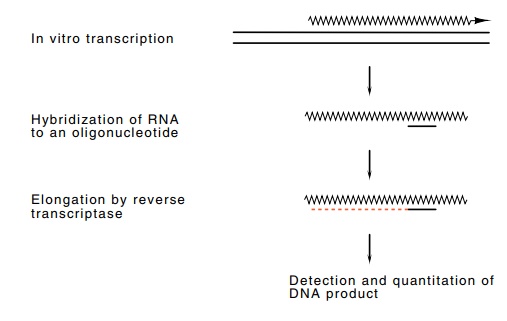
Figure 4.4 Assay for the presence of an RNA sequence by elongation withreverse transcriptase.
Assaying
transcription from an in vitro
reaction is relatively easy if purified proteins can be used. Obtaining the
purified proteins is not so easy, however, since often their very purification
requires assaying crude extracts for the transcriptional activity. The presence
of nucleo-tides in the extracts precludes radioactive labeling of the RNA
synthe-sized in vitro. Therefore an
assay is required that can utilize nonradioactive RNA. Primer extension meets
the requirements for such an assay (Fig. 4.4). RNA that was initiated in vitro and which transcribed
off the
end of a DNA template is purified away from proteins and some of the DNA. This
RNA is hybridized to a short DNA fragment. Then reverse transcriptase, an
enzyme utilized by RNA viruses to make DNA copies of themselves, and
radioactive deoxynucleotide triphosphates are added. The reverse transcriptase
synthesizes a radioactive DNA copy of the RNA.
This can be separated from extraneous and nonspecific DNA fragments by
electrophoresis and be quantitated by autoradiography.
Related Topics