Chapter: Clinical Anesthesiology: Anesthetic Management: Cardiovascular Physiology & Anesthesia
Determinants of Ventricular Performance
DETERMINANTS OF VENTRICULAR PERFORMANCE
Discussions of ventricular function
usually refer to the left ventricle, but the same concepts apply to the right
ventricle. Although the ventricles are often thought of as functioning
separately, their interde-pendence has clearly been demonstrated. Moreover,
factors affecting systolic and diastolic functions can be differentiated: Systolic
function involves ventric-ular ejection, whereas diastolic function is related
to ventricular filling.
Ventricular systolic function is often
(errone-ously) equated with cardiac output, which can be defined as the volume
of blood pumped by the heart per minute. Because the two ventricles function in
series, their outputs are normally equal. Cardiac out-put (CO) is expressed by
the following equation:
CO = SV × HR
where SV is the stroke volume (the
volume pumped per contraction) and HR is heart rate. To compensate for
variations in body size, CO is often expressed in terms of total body surface
area:
where CI is the cardiac index and BSA is
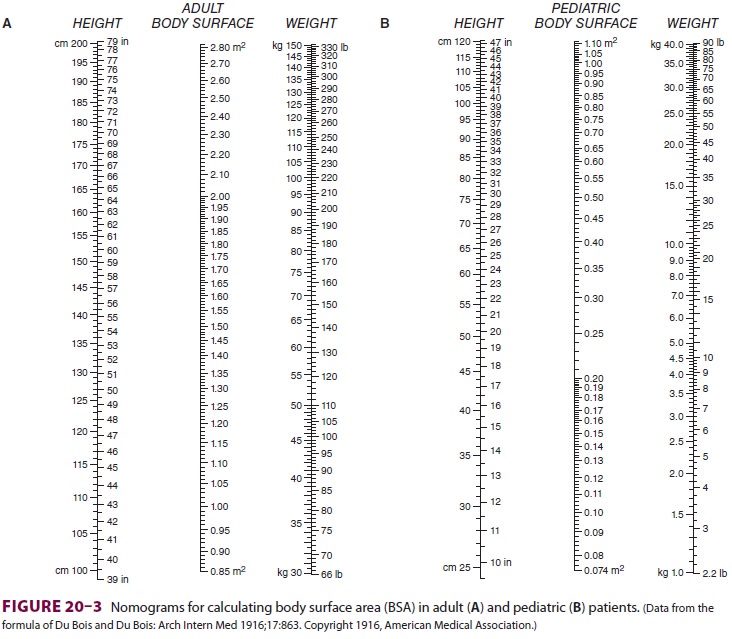
body sur-face area. BSA is usually obtained from nomograms
based on height and weight (Figure 20–3).
Normal CI is 2.5–4.2 L/min/m2. Because the normal CI has a wide
range, it is a relatively insensitive measurement of ventricular performance.
Abnormalities in CI therefore usually reflect gross ventricular impairment. A more
accurate assess-ment can be obtained if the response of the cardiac output to
exercise is evaluated. Under these condi-tions, failure of the cardiac output
to increase and keep up with oxygen consumption is reflected by a decreasing
mixed venous oxygen saturation. A decrease in mixed venous oxygen saturation in
response to increased demand usually reflects inadequate tissue perfusion.
Thus, in the absence of hypoxia or severe anemia, measurement of mixed venous
oxygen tension (or satura-tion) is an excellent estimate of the adequacy of
cardiac output.
1. Heart Rate
When stroke volume remains constant,
cardiac out-put is directly proportional to heart rate. Heart rate is an
intrinsic function of the SA node (spontane-ous depolarization), but is
modified by autonomic, humoral, and local factors. The normal intrinsic rate of
the SA node in young adults is about 90– 100 beats/min, but it decreases with
age based on the following formula:
Normal intrinsic heart rate = 118 beats/min − (0.57 × age)
Enhanced vagal activity slows the heart
rate via stimulation of M 2 cholinergic receptors, whereas enhanced
sympathetic activity increases the heart rate mainly through activation of β1-adrenergic receptors and, to lesser
extent, β2-adrenergic
recep-tors (see above).
2. Stroke Volume
Stroke volume is normally determined by
three major factors: preload, afterload, and contractility. This analysis is
analogous to laboratory observa-tions on skeletal muscle preparations. Preload
is muscle length prior to contraction, whereas after-load is the tension
against which the muscle must
contract. Contractility is an intrinsic
property of the muscle that is related to the force of contrac-tion but is
independent of both preload and after-load. Because the heart is a
three-dimensional multichambered pump, both ventricular geometric form and
valvular dysfunction can also affect stroke volume (Table 20–3).
Preload
Ventricular preload is end-diastolic
volume, which is generally dependent on ventricular filling. The relationship
between cardiac output and left ven-tricular end-diastolic volume is known as
Starling’s law of the heart (Figure 20–4). Note that when the heart rate and
contractility remain constant, car-diac output is directly proportional to
preload until excessive end-diastolic volumes are reached. At that
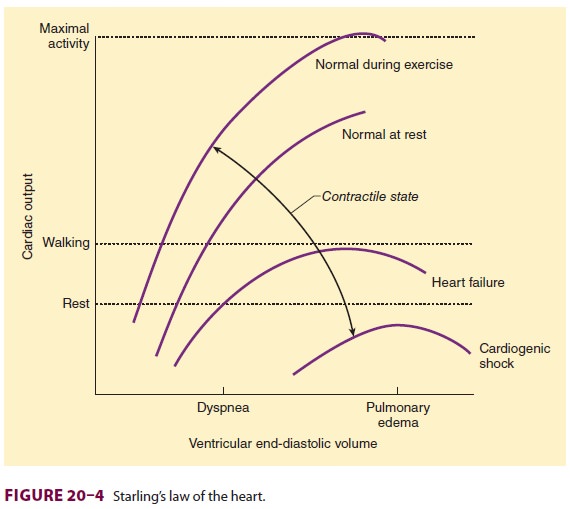
point, cardiac output does not
appreciably change— or may even decrease. Excessive distention of either
ventricle can lead to excessive dilatation and incom-petence of the AV valves.
A. Determinants of Ventricular Filling
Ventricular fi lling can be influenced
by a variety of factors (Table 20–4), of which the most impor-tant is
venous return. Because most of the other
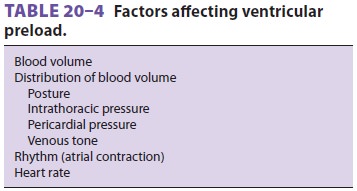
factors affecting venous return are
usually fixed, vascular capacity is normally its major determi-nant. Increases
in metabolic activity reduce vas-cular capacity, so that venous return to the
heart increases as the volume of venous capacitance vessels decreases. Changes
in blood volume and venous tone are important causes of intraopera-tive and
postoperative changes in ventricular fill-ing and cardiac output. Any factor
that alters the normally small venous pressure gradient favoring blood return
to the heart also affects cardiac filling. Such factors include changes in
intrathoracic pres-sure (positive-pressure ventilation or thoracotomy), posture
(positioning during surgery), and pericar-dial pressure (pericardial disease).
The most important determinant of right
ven-tricular preload is venous return. In the absence of significant pulmonary
or right ventricular dys-function, venous return is also the major deter-minant
of left ventricular preload. Normally, the end-diastolic volumes of both
ventricles are similar,and, normally, the venous return is numerically
equivalent to the cardiac output.
Both heart rate and rhythm can also
affect ven-tricular preload. Increases in heart rate are associ-ated with
proportionately greater reductions in diastole than systole. Ventricular
filling therefore progressively becomes impaired at increased heart rates (>120 beats/min in adults). Absent (atrial
fibrillation), ineffective (atrial flutter), or altered timing of atrial
contraction (low atrial or junctional rhythms) can also reduce ventricular
filling by 20% to 30%. Patients with reduced ventricular compliance are more
affected by the loss of anormally timed atrial systole than are those with
normal ventricular compliance.
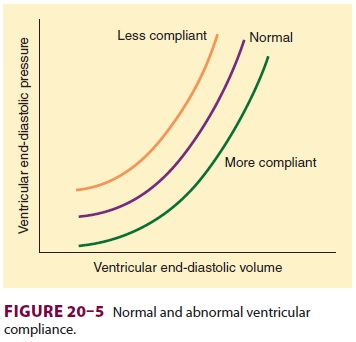
B. Diastolic Function and Ventricular Compliance
Left ventricular end-diastolic pressure
(LVEDP) can be used as a measure of preload only if the relationship between
ventricular volume and pressure (ventricular compliance) is constant. However,
ventricular compliance is normally non-linear (Figure
20–5). Impaired diastolic function reduces ventricular compliance.
Therefore, the same LVEDP that corresponds to a normal preload in a normal
patient may correspond to a decreased pre-load in a patient with impaired
diastolic function.
Many factors are known to influence
ventricular diastolic function and compliance. Nonetheless, measurement of
LVEDP or other pressures approx-imating LVEDP (such as pulmonary artery
occlu-sion pressure) are potential means of estimating left ventricular
preload. Changes in central venous pressure can be used as a rough index for
changes in right and left ventricular preload in most normal individuals.
Factors affecting ventricular compliance
can be separated into those related to the rate of relax-ation (early diastolic
compliance) and passive stiffness of the ventricles (late diastolic
compli-ance). Hypertrophy (from hypertension or aortic valve stenosis),
ischemia, and asynchrony reduce early compliance; hypertrophy and fibrosis
reduce late compliance. Extrinsic factors (such as pericar-dial disease,
excessive distention of the contralat-eral ventricle, increased airway or
pleural pressure, tumors, and surgical compression) can also reduce ventricular
compliance. Because of its normally thinner wall, the right ventricle is more
compliant than the left.
Afterload
Afterload for the intact heart is
commonly equated with either ventricular wall tension during systole or arterial
impedance to ejection. Wall tension may be thought of as the pressure the
ventricle must over-come to reduce its cavity volume. If the ventricle is
assumed to be spherical, ventricular wall tension can be expressed by Laplace’s
law:

where P is intraventricular pressure, R
is the ven-tricular radius, and H is
wall thickness. Although the normal ventricle is usually ellipsoidal, this
relationship is still useful. The larger the ventricu-lar radius, the greater
the wall tension required to develop the same ventricular pressure. Conversely,
an increase in wall thickness reduces ventricular wall tension.
Systolic intraventricular pressure is
dependent on the force of ventricular contraction; the visco-elastic properties
of the aorta, its proximal branches,
and blood (viscosity and density); and systemicvascular resistance (SVR).
Arteriolar tone is theprimary determinant of SVR. Because viscoelastic
properties are generally fixed in any given patient, left ventricular afterload
is usually equated clini-cally with SVR, which is calculated by the following
equation:
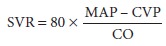
where MAP is mean arterial pressure in
millime-ters of mercury, CVP is central venous pressure in millimeters of
mercury, and CO is cardiac output in liters per minute. Normal SVR is 900–1500
dyn · s cm–5. Systolic blood pressure may also be
used as an approximation of left ventricular afterload in the absence of
chronic changes in the size, shape, or thickness of the ventricular wall or
acute changes in systemic vascular resistance. Some clinicians prefer to use CI
instead of CO in calculating a systemic vas-cular resistance index (SVRI), so
that SVRI = SVR × BSA.
Right ventricular afterload is mainly
depen-dent on pulmonary vascular resistance (PVR) and is expressed by the
following equation:
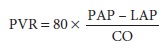
where PAP is mean pulmonary artery
pressure and LAP is left atrial pressure. In practice, pulmonary capillary
wedge pressure (PCWP) is usually substi-tuted as an approximation for LAP.
Normal PVR is 50–150 dyn · s cm–5.
Cardiac output is inversely related to
large changes in afterload on the left ventricle; however, small increases or
decreases in afterload may have no effect at all on cardiac output. Because of
its thinner wall, the right ventricle is more sensitive to changes in afterload
than is the left ventricle.Cardiac output in patients with marked right or left
ventricular impairment is verysensitive to acute increases in afterload. The
latter is particularly true in the presence of drug- or ischemia-induced
myocardial depression or chronic heart failure.
Contractility
Cardiac contractility (inotropy) is the
intrinsic ability of the myocardium to pump in the absence of changes in
preload or afterload. Contractility is related to the rate of myocardial muscle
shortening, which is, in turn, dependent on the intracellular Ca2+ concentration during systole. Increases in heart
rate can also enhance contractility under some condi-tions, perhaps because of
the increased availability of intracellular Ca 2+.
Contractility can be altered by neural,
humoral, or pharmacological influences. Sympathetic nervous system activity
normally has the most important effect on contractility. Sympathetic fibers
innervate atrial and ventricular muscle, as well as nodal tissues. In addition
to its positive chronotropic effect, norepi-nephrine release also enhances
contractility primar-ily via β1-receptor activation. α-Adrenergic receptors are also present
in the myocardium, but seem to have only minor positive inotropic and
chronotropic effects. Sympathomimetic drugs and secretion of epi-nephrine from
the adrenal glands similarly increase contractility via β1-receptor activation.
Myocardial contractility is depressed by
hypoxia, acidosis, depletion of catecholamine stores within the heart, and loss
of functioning muscle mass as a result of ischemia or infarction. At large
enough doses, most anesthetics and antiarrhyth-mic agents are negative
inotropes (ie, they decrease contractility).
Wall Motion Abnormalities
Regional wall motion abnormalities cause
a break-down of the analogy between the intact heart and skeletal muscle
preparations. Such abnormalities may be due to ischemia, scarring, hypertrophy,
or altered conduction. When the ventricular cavity does not collapse
symmetrically or fully, emptying becomes impaired. Hypokinesis (decreased
contrac-tion), akinesis (failure to contract), and dyskinesis (paradoxic
bulging) during systole reflect increasing degrees of contraction abnormalities.
Although con-tractility may be normal or even enhanced in some areas,
abnormalities in other areas of the ventricle can impair emptying and reduce
stroke volume. The severity of the impairment depends on the size and number of
abnormally contracting areas.
Valvular Dysfunction
Valvular dysfunction can involve any one
of the four valves in the heart and can include stenosis, regur-gitation
(incompetence), or both. Stenosis of an AV valve (tricuspid or mitral) reduces
stroke volume primarily by decreasing ventricular preload, whereas stenosis of
a semilunar valve (pulmonary or aortic) reduces stroke volume primarily by
increasing ven-tricular afterload. In contrast, valvular regurgita-tion can
reduce stroke volume without changes in preload, afterload, or contractility
and without wall motion abnormalities. The effective stroke volume is reduced
by the regurgitant volume with every con-traction. When an AV valve is
incompetent, a sig-nificant part of the ventricular end-diastolic volume can
flow backward into the atrium during systole; the stroke volume is reduced by
the regurgitant vol-ume. Similarly, when a semilunar valve is incompe-tent, a
fraction of end-diastolic volume arises from backward flow into the ventricle
during diastole.
Related Topics