Chapter: Clinical Anesthesiology: Anesthetic Management: Cardiovascular Physiology & Anesthesia
Compensatory Mechanisms
COMPENSATORY MECHANISMS
Compensatory mechanisms generally
present in patients with heart failure include increased pre-load, activation
of the sympathetic nervous system and the renin–angiotensin–aldosterone system,
and increased release of AVP. Although these mechanisms can initially
compensate for mild to moderate cardiac dysfunction, with increasing severity
of dysfunction, they may actually contrib-ute to the cardiac impairment. Many
of the drug treatments of chronic heart failure serve to coun-teract these
mechanisms.
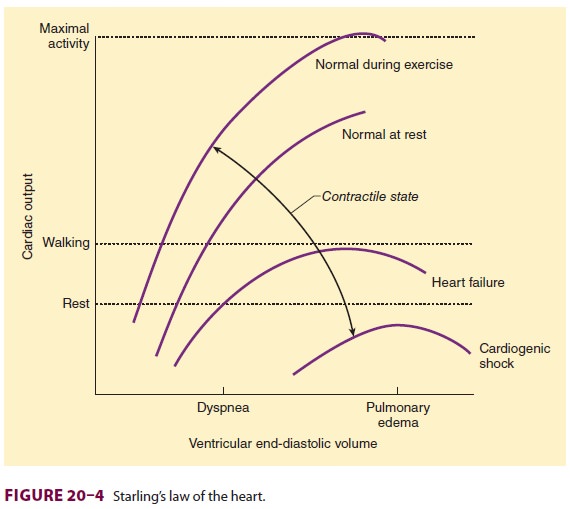
Increased Preload
An increase in ventricular size not only
reflects an inability to keep up with an increased circulating blood volume,
but also serves to increase stroke vol-ume by moving the heart up the Starling
curve (see Figure 20–4). Even when EF is reduced, an increase in ventricular
end-diastolic volume can maintain a normal stroke volume. Worsening venous conges-tion
caused by the pooling of blood behind the fail-ing ventricle and excessive
ventricular dilatation can rapidly lead to clinical deterioration. Left
ventricu-lar failure results in pulmonary vascular congestion and progressive
transudation of fluid, first into the pulmonary interstitium and then into
alveoli (pul-monary edema). Right ventricular failure leads to systemic venous
hypertension, which results in peripheral edema, hepatic congestion and
dysfunc-tion, and ascites. Dilatation of the annulus of either AV valve leads
to valvular regurgitation, further impairing ventricular output.
Increased Sympathetic Tone
Sympathetic activation increases release
of norepi-nephrine from nerve endings in the heart and the adrenal secretion of
epinephrine into the circula-tion. Plasma catecholamine levels are generally
directly proportional to the degree of left ventricu-lar dysfunction. Although
enhanced sympathetic outflow can initially maintain cardiac output by
increasing heart rate and contractility, worsening ventricular function elicits
increasing degrees of vasoconstriction in an effort to maintain arterial blood
pressure. The associated increase in afterload, however, reduces cardiac output
and exacerbates the ventricular failure.Chronic sympathetic activation in
patients with heart failure eventually decreases the response of adrenergic
receptorsto catecholamines (receptor uncoupling), the number of receptors
(down-regulation), and cardiac catecholamine stores. 11 Nonetheless, the
failing heart becomesincreasingly dependent on circulating cate-cholamines.
Abrupt withdrawal in sympathetic out-flow or decreases in circulating
catecholamine levels, such as can occur following induction of anesthesia, may
lead to acute cardiac decompensa-tion. A reduced density of M 2 receptors also decreases parasympathetic influences
on the heart.
Sympathetic activation tends to
redistribute systemic blood flow output away from the skin, gut, kidneys, and
skeletal muscle to the heart and brain. Decreased renal perfusion, together
with β1-adrenergic
activity at the juxtaglomerular appa-ratus, activates the
renin–angiotensin–aldosterone axis, which leads to sodium retention and
intersti-tial edema. Moreover, vasoconstriction secondary to elevated
angiotensin II levels increases left ven-tricular afterload and causes further
deterioration of systolic function. The latter partially accounts for the
efficacy of angiotensin-converting enzyme (ACE) inhibitors and angiotensin
receptor block-ers in heart failure. Symptoms may also improve in some patients
with careful, low-dose β-adrenergic blockade. Outcomes in heart
failure are improved by administration of ACE inhibitors (and/or angiotensin
receptor blockers), certain long-acting β-blockers (carvedilol or extended
release meto-prolol), and aldosterone inhibitors (spironolactone or
eplerenone).Circulating AVP levels, often markedly increased in patients with
severe heart failure, will increase ventricular afterload and are responsible
for a defect in free water clearance that is commonly associated with
hyponatremia.
Brain natriuretic peptide (BNP) is
produced in the heart in response to myocyte distention. Elevated BNP
concentration (>500 pg/mL) usually indicates heart
failure, and measurement of BNP concentration can be used to distinguish
between heart failure and lung disease as a cause of dyspnea. Recombinant BNP
was developed as a vasodilator and inhibitor of the
renin–angiotensin–aldoste-rone system for use in patients with severe
decom-pensated heart failure, but outcomes were not improved with its use.
Ventricular Hypertrophy
Ventricular hypertrophy can occur with
or without dilatation, depending on the type of stress imposed on the
ventricle. When the heart is subjected to either pressure or volume overload,
the initial response is to increase sarcomere length and optimally overlap
actin and myosin. With time, ventricular muscle mass begins to increase in
response to the abnormal stress.
In the volume-overloaded ventricle, the
prob-lem is an increase in diastolic wall stress. The increase in ventricular
muscle mass is sufficient only to compensate for the increase in diameter: The
ratio of the ventricular radius to wall thickness is unchanged. Sarcomeres
replicate mainly in series, resulting in eccentric hypertrophy. Although ven-tricular
EF remains depressed, the increase in end-diastolic volume can maintain normal
at-rest stroke volume (and cardiac output).
The problem in a pressure-overloaded
ven-tricle is an increase in systolic wall stress. In this case, sarcomeres
mainly replicate in parallel, result-ing in concentric hypertrophy: The
hypertrophy is such that the ratio of myocardial wall thickness to ventricular
radius increases. As can be seen from Laplace’s law, systolic wall stress can
then be nor-malized. Ventricular hypertrophy, particularly that caused by
pressure overload, usually results in pro-gressive diastolic dysfunction. The
most common reasons for isolated left ventricular hypertrophy are hypertension
and aortic stenosis.
Related Topics