Chapter: Modern Pharmacology with Clinical Applications: Pharmacological Management of Chronic Heart Failure
Cardiac Electrophysiology: Triggered Activity
Triggered Activity
Triggered activity occurs
when after-depolarizations in-duced by a preceding action potential raise the
resting membrane potential above the threshold value, leading to an additional
action potential. After-depolarizations may be categorized as early, occurring
during phase III of the action potential before achieving full repolarization,
or delayed, occurring after full repolarization of the membrane.
After-depolarizations may stimulate an iso-lated extrapropagated impulse or
lead to sustained repet-itive activity. The crucial difference between
triggered ac-tivity and abnormal automaticity is that triggered activity
depends on a preceding action potential and cannot be self-induced.
After-depolarizations or triggered activity are often associated with excessive
increases in intracel-lular [Ca ]. The potential for development of triggered
activity is accentuated in the presence of an increase in extracellular [Ca ]
that would increase the amount of ionized calcium entering the cell during
depolarization. Furthermore, conditions or pharmacological interven-tions
favoring prolongation of the plateau (phase 3) of the action potential and
prolongation of the QT interval of the electrocardiogram would increase
intracellular [Ca++ ] and the potential for proarrhythmia.
Early after-depolarizations
are purported to be the mechanism giving rise to torsades de pointes. Conditions or drugs known to prolong the action
potential, espe-cially by interventions that decrease the outward potas-sium
currents, facilitate development of torsades de pointes tachyarrhythmias. Early
after-depolarizations may develop in association with hypokalemia, hypoxia,
acidosis, and a wide range of pharmacological agents that interfere with
outward currents or enhance inward currents. Antiarrhythmic agents, in
particular sotalol, quinidine, and dofetilide, may give rise to
after-depolar-izations and torsades de pointes tachyarrhythmia in persons with
underlying cardiac abnormalities or alter-ations in plasma electrolytes.
Conditions leading to bradycardia also may facilitate development of torsades
de pointes tachyarrhythmia.
Early after-depolarizations
and the associated ven-tricular arrhythmia can be prevented or suppressed by
the appropriate adjustment of plasma potassium and/or magnesium concentrations.
Lidocaine or procainamide may be effective for termination of the arrhythmia.
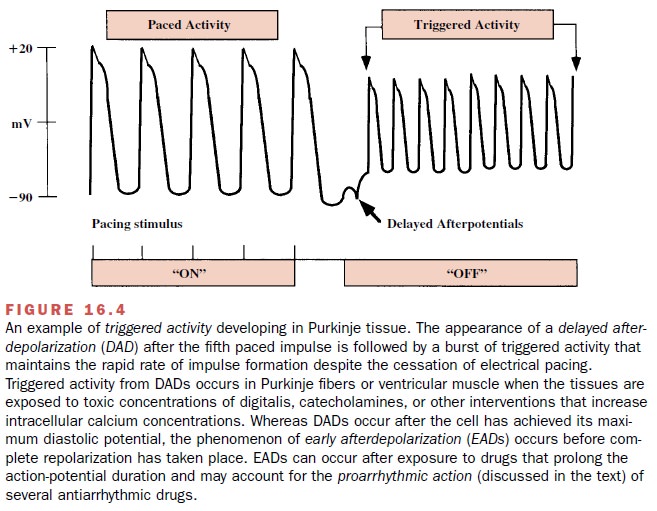
Delayed after-depolarizations
(Figure 16.4) may oc-cur in the presence of a rapid heart rate, digitalis
glyco-sides, hypokalemia, hypercalcemia and catecholamines. Each of these
influences ultimately leads to an increase in intracellular ionized calcium
that is known to activate an inward ionic current. The inward ionic current
acti-vates a nonselective channel that normally is involved with the transport
of sodium but that under pathophys-iological conditions may permit the movement
of sodium or potassium ions. Upon reaching threshold, the calcium-induced oscillatory
potentials lead to the pro-duction of a sustained ventricular arrhythmia.
Delayed after-depolarizations, in contrast to early after-depolar-izations, are
more likely to produce triggered tachy-arrhythmias during periods of short
pacing cycle lengths (rapid heart rates). Exercise-induced ventricular
tachy-cardia in persons without overt cardiac disease exem-plifies such a
situation. The electrophysiological abnor-mality is catecholamine dependent and
calcium sensitive. The arrhythmia may respond to L-type cal-cium channel
antagonists or inhibitors of the cardiac β-adrenoceptor. Each of these approaches would
serve to reduce the tissue calcium concentration.
Related Topics