Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Pharmacokinetics and Pharmacodynamics of Peptide and Protein Drugs
Absorption of Protein Therapeutics - Pharmacokinetics of Protein Therapeutics
Absorption of Protein Therapeutics
Enteral Administration
Peptides and proteins, unlike conventional small molecule drugs, are generally not therapeutically active upon oral administration (Fasano, 1998; Mahato et al., 2003; Tang et al., 2004). The lack of systemic bioavailability is mainly caused by two factors: (1) high gastrointestinal enzyme activity and (2) low permeability through the gastrointestinal mucosa. In fact, the substantial peptidase and protease activity in the gastrointestinal tract makes it the most efficient body compartment for peptide and protein metabo-lism. Furthermore, the gastrointestinal mucosa pre-sents a major absorption barrier for water-soluble macromolecules such as peptides and proteins (Tang et al., 2004). Thus, although various factors such as permeability, stability and gastrointestinal transit time can affect the rate and extent of absorption of orally administered proteins, molecular size is generally considered the ultimate obstacle (Shen, 2003).
Since oral administration is still a highly desir-able route of delivery for protein drugs due to its convenience, cost-effectiveness and painlessness, numerous strategies to overcome the obstacles asso-ciated with oral delivery of proteins have recently been an area of intensive research. Suggested approaches to increase the oral bioavailability of protein drugs include encapsulation into micro- or nanoparticles thereby protecting proteins from intest-inal degradation (Lee, 2002; Mahato et al., 2003; Shen, 2003). Other strategies are chemical modifications such as amino acid backbone modifications and chemical conjugations to improve the resistance to degradation and the permeability of the protein drug. Coadministration of protease inhibitors has also been suggested for the inhibition of enzymatic degradation (Pauletti et al., 1997; Mahato et al., 2003). More details on approaches for oral delivery of peptide and protein therapeutics are discussed.
Parenteral Administration
Most peptide and protein drugs are currently for-mulated as parenteral formulations because of their poor oral bioavailability. Major routes of administra-tion include intravenous (IV), subcutaneous (SC), and intramuscular (IM) administration. In addition, other non-oral administration pathways are utilized, in-cluding nasal, buccal, rectal, vaginal, transdermal, ocular and pulmonary drug delivery.
IV administration of peptides and proteins offers the advantage of circumventing presystemic degrada-tion, thereby achieving the highest concentration in the biological system. Protein therapeutics given by the IV route include, among many others, the tissue plasminogen activator (t-PA) analogs alteplase and tenecteplase, the recombinant human erythropoietin epoetin-a, and the granulocyte-colony-stimulating factor filgrastim (Tang and Meibohm, 2006).
IV administration as either a bolus dose or constant rate infusion, however, may not always provide the desired concentration–time profile de-pending on the biological activity of the product. In these cases, IM or SC injections may be more appro-priate alternatives. For example, luteinizing hormone-releasing hormone (LH-RH) in bursts stimulates the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH), whereas a continuous base-line level will suppress the release of these hormones (Handelsman and Swerdloff, 1986). To avoid the high peaks from an IV administration of leuprorelin, an LH-RH agonist, a long acting monthly depot injection of the drug is approved for the treatment of prostate cancer and endometriosis (Periti et al., 2002). A recent study comparing SC versus IV administration of epoetin-a in patients receiving hemodialysis reports that the SC route can maintain the hematocrit in a desired target range with a lower average weekly dose of epoetin-a compared to IV (Kaufman et al., 1998).
One of the potential limitations of SC and IM administration, however, are the presystemic degra-dation processes frequently associated with these administration routes, resulting in a reduced bioavail-ability compared to IV administration. The pharma-cokinetically derived apparent absorption rate constant kapp for protein drugs administered via these administration routes is thus the combination of absorption into the systemic circulation and presys-temic degradation at the absorption site, i.e., the sum of a true first-order absorption rate constant ka and a first-order degradation rate constant. The true absorp-tion rate constant ka can then be calculated as
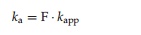
where F is the bioavailability compared to IV administration. A rapid apparent absorption, i.e., large kapp, can thus be the result of a slow true absorption and a fast presystemic degradation, i.e., a low systemic bioavailability (Colburn, 1991).
Other potential factors that may limit bioavail-ability of proteins after SC or IM administration include variable local blood flow, injection trauma, and limitations of uptake into the systemic circulation related to effective capillary pore size and diffusion.
Several peptide and protein therapeutics including anakinra, etanercept, insulin, and pegfilgrastim are administered as SC injections. Following an SC injection, peptide and protein therapeutics may enter the systemic circulation either via blood capillaries or through lymphatic vessels (Porter and Charman, 2000). In general, macromolecules larger than 16 kDa are pre-dominantly absorbed into the lymphatics whereas those under 1 kDa are mostly absorbed into the blood circulation. There appears to be a linear relationshipbetween the molecular weight of the protein and the proportion of the dose absorbed by the lymphatics (see Fig. 12) (Supersaxo et al., 1990).
This is of particular importance for those agents whose therapeutic targets are lymphoid cells (i.e., interferons and interleukins). Studies with recombi-nant human interferon a-2a (rhIFNα2a) indicate that following SC administration, high concentrations of the recombinant protein are found in the lymphatic system, which drains into regional lymph nodes (Supersaxo et al., 1988). Clinical studies show that palliative low-to-intermediate-dose SC recombinant interleukin-2 (rIL-2) in combination with rhIFNα2a can be administered to patients in the ambulatory setting with efficacy and safety profiles comparable to the most aggressive IV rIL-2 protocol against meta-static renal cell cancer (Schomburg et al., 1993; Chen et al., 2000).
Related Topics