Chapter: Genetics and Molecular Biology: Genetic Engineering and Recombinant DNA
Chemical DNA Sequencing
Chemical DNA Sequencing
Two techniques were developed for DNA sequencing:
chemical and enzymatic. Initially more DNA was sequenced with the chemical
method, but improvements in the enzymatic method now make it the method of
choice for almost all sequencing problems. The application of the chemical
methods, however, are highly useful for biochemical studies of protein-DNA
interactions, and therefore also find significant use. Both the chemical and
the enzymatic sequencing methods utilize electrophoresis at high temperatures
in the presence of urea so as to denature the DNA. Under these conditions the
single-stranded mole-cules migrate at rates almost independent of their
sequence and depend-ent only on their length. In such gels two single-stranded
DNA fragments differing in length by a single base can be resolved from one
another if their lengths are less than 300 to 500 bases.
The basic principle of DNA sequencing by the
chemical or enzymatic method is to generate a set of radioactive DNA fragments
covering a region. The sizes of these fragments indicate the nucleotide
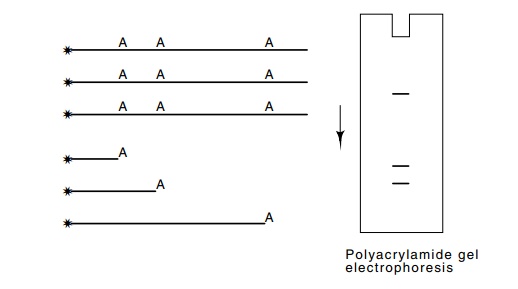
sequence of the region. How is this possible? Consider a large number of
single-stranded molecules (Fig. 9.16). Suppose the 5’ ends of all the molecules
are at the same nucleotide. Suppose also that the 3’ ends of some of the
molecules in the population are at the first A residue after the 5’ end, some
end at the second A residue and so on. That is, the population of molecules
consists of some ending at each of the A’s. If the first few A
Figure
9.16 DNA sequencing. Modification at a
average of one A per moleculein the population followed by cleavage at the
modification sites generates a population of molecules ending at the former
positions of A’s. Their electropho-resis generates the ladder pattern
indicated.
In the same way that A-specific ends could be used
to determine the distances of A residues from the end, other base-specific
terminations could be used to determine the distances of the other nucleotides
from the 5’ end.
Practical considerations slightly complicate the
above procedure. First, the amounts of DNA that must be identified are too
small to be detected by staining with ethidium bromide and observing the
fluores - cent DNA bands. Second, the chemical method generates extraneous
fragments in addition to those originating from the one end under
consideration. These other fragments could interfere with the identifi-cation
of the desired fragments. Both problems are solved by radioac-tively labeling
the DNA at its 5’ end. Autoradiography of the gels then provides adequate sensitivity,
and only the fragments that are radioac-tive and generate bands on the
autoradiographs are precisely those that include the labeled end.
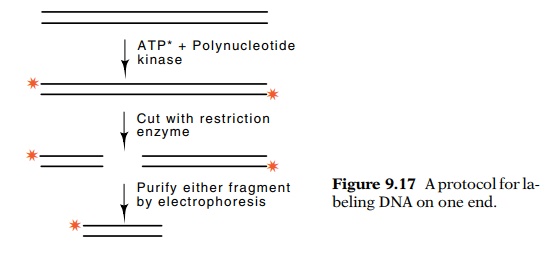
Obtaining a DNA fragment labeled only at one end is
not difficult. Each strand in a double-stranded segment can be labeled on its
5’ end with the enzyme T4 phage polynucleotide kinase. Then the two strands can
be separated by denaturation of the DNA and electrophoresis under partially
denaturing conditions. Frequently the two strands migrate at different rates and
can be separated. If so, either can be used in chemical sequencing. A second
way to obtain the DNA fragment with a radioac-tive label on only one end also
begins with end-labeled double-stranded duplex (Fig. 9.17). This duplex is next
cleaved with a restriction enzyme and if the two fragments are unequal in size,
they can be separated by electrophoresis. Either of the fragments is suitable
for DNA sequencing because only one of the strands in each fragment is then
radioactive, and it is labeled on only one end.
To sequence a stretch of DNA, four populations of
radiolabeled DNA are made, one partially cleaved at each of the four bases.
These four populations are then subjected to electrophoresis in four adjacent
lanes
Figure
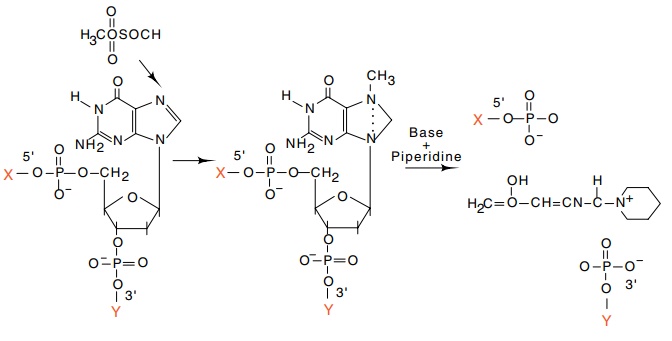
9.18 Basis of the G-specific reaction
in Maxam-Gilbert sequencing. Thefinal result is strand scission at the former
position of a guanosine.
In the chemical sequencing method the cleavages at
the four bases are made by subjecting the labeled DNA to conditions that
generate base-specific cleavage of the phosphodiester bonds. An average of
about one cleavage per several hundred bases is optimal for most sequencing.
Maxam and Gilbert discovered that to generate the requisite base specificity,
the procedure needed to be broken into two parts. The first part introduces a
highly base-specific chemical modification under controlled and mild
conditions. Then harsh conditions are used to generate the actual cleavages at
all the modified positions.
Dimethylsulfate provides a highly specific
methylation of guanines (Fig. 9.18). The introduction of the methyl group
permits subsequent depurination followed by cleavage of the sugar-phosphate
backbone with piperidine. Slightly altered conditions yield methylation by
di-methyl sulfate of guanines, and to a lesser extent, methylation of adenines.
Hydrazine is used for pyrimidine-specific reactions. Both thymine and cytosine
react with hydrazine unless high concentrations of salt are present. Then only
cytosine reacts. These reactions yield bands for G’s, G’s + A’s, C’s + T’s, and
T’s. These are sufficient for sequence determination.
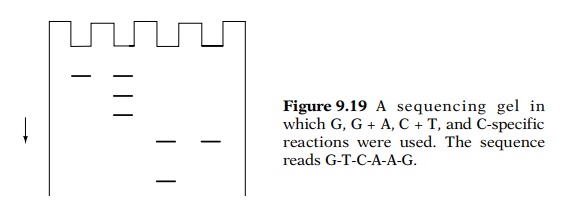
Consider, for example, that the DNA sequence near
the 5’ end of the fragment was 5’-GTCAAG-3’ and the fragment was labeled on its
5’ end. Then electrophoresis of the four reaction products yields a ladder
pattern from which the sequence may be read by proceeding upward on the gel
from lane to lane (Fig. 9.19). Sequences of up to about 200 bases may be read
from a single set of reactions.
Related Topics