Chapter: Basic & Clinical Pharmacology : Drugs Used in Disorders of Coagulation
Warfarin & Other Coumarin Anticoagulants
WARFARIN & OTHER COUMARIN
ANTICOAGULANTS
Chemistry & Pharmacokinetics
The
clinical use of the coumarin anticoagulants began with the discovery of an
anticoagulant substance formed in spoiled sweet clover silage which caused
hemorrhagic disease in cattle. At the behest of local farmers, a chemist at the
University of Wisconsin identified the toxic agent as bishydroxycoumarin. A
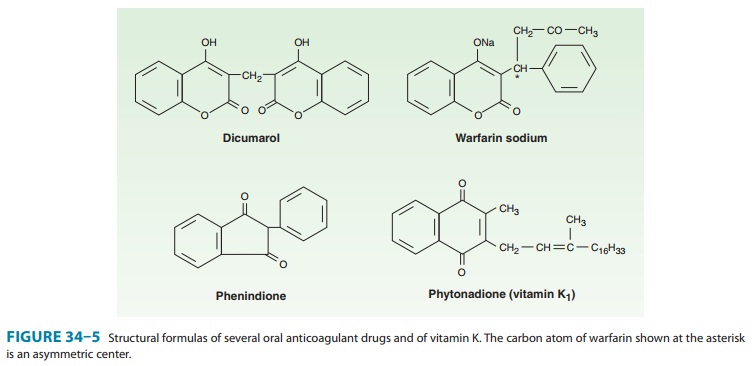
synthesized derivative, dicumarol and its congeners, most notably warfarin (Wisconsin Alumni Research Foundation, with “arin” from cou-marin
added; Figure 34–5), were initially used as rodenticides. In the 1950s warfarin
(under the brand name Coumadin) was introduced as an antithrombotic agent in
humans. Warfarin is one of the most commonly prescribed drugs, used by
approxi-mately 1.5 million individuals, and several studies have indicated that
the drug is significantly underused in clinical situations where it has proven
benefit.
Warfarin is generally administered as the sodium salt and has 100% bioavailability. Over 99% of racemic warfarin is bound to plasma albumin, which may contribute to its small volume of dis-tribution (the albumin space), its long half-life in plasma (36 hours), and the lack of urinary excretion of unchanged drug. Warfarin used clinically is a racemic mixture composed of equal amounts of two enantiomorphs.
The
levorotatory S-warfarin is four times
more potent than the dextrorotatory R-warfarin.
This observation is use-ful in understanding the stereoselective nature of
several drug inter-actions involving warfarin.
Mechanism of Action
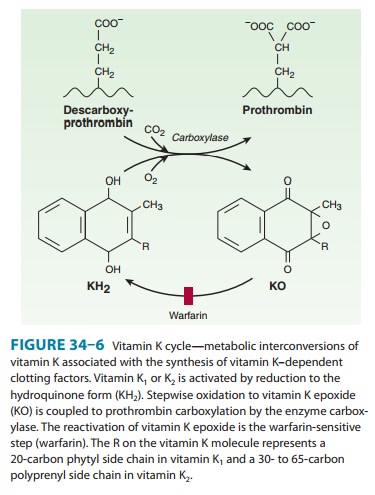
Coumarin
anticoagulants block the γ-carboxylation of several glutamate
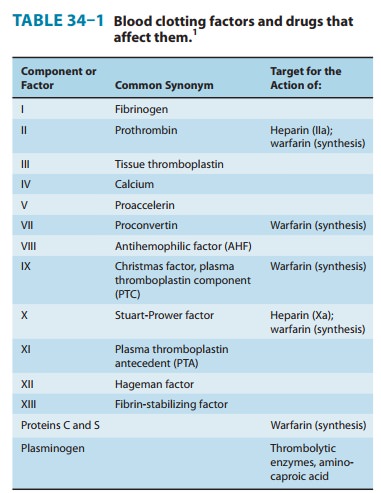
residues in prothrombin and factors VII, IX, and X as well as the endogenous
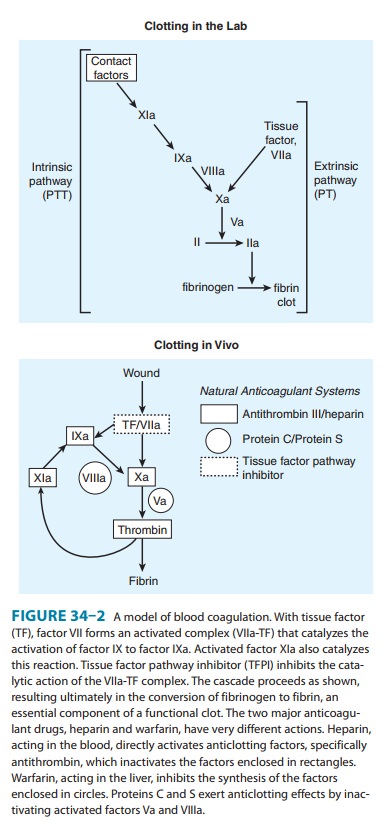
anticoagulant proteins C and S (Figure 34–2, Table 34–1). The blockade results
in incomplete coagulation factor molecules that are biologically inactive. The
protein carboxylation reaction is coupled to the oxidation of vitamin K. The
vitamin must then be reduced to reactivate it. Warfarin prevents reductive metabolism
of the inactive vitamin K epoxide back to its active hydroquinone form (Figure
34–6). Mutational change of the responsible enzyme, vitamin K epox-ide
reductase, can give rise to genetic resistance to warfarin in humans and
especially in rats.
There
is an 8- to 12-hour delay in the action of warfarin. Its anticoagulant effect
results from a balance between partially inhib-ited synthesis and unaltered
degradation of the four vitamin K-dependent clotting factors. The resulting
inhibition of coagula-tion is dependent on their degradation half-lives in the
circulation. These half-lives are 6, 24, 40, and 60 hours for factors VII, IX,
X, and II, respectively. Larger initial doses of warfarin—up to about 0.75
mg/kg—hasten the onset of the anticoagulant effect. Beyond this dosage, the
speed of onset is independent of the dose size. The only effect of a larger
loading dose is to prolong the time that the plasma concentration of drug
remains above that required for sup-pression of clotting factor synthesis. The
only difference among oral anticoagulants in producing and maintaining
hypoprothrom-binemia is the half-life of each drug.
Toxicity
Warfarin crosses the
placenta readily and can cause a hemorrhagic disorder in the fetus.
Furthermore, fetal proteins with γ-carboxyglutamate residues found in bone and
blood may beaffected by warfarin; the drug can cause a serious birth defect
characterized by abnormal bone formation. Thus, warfarin should never be administered
during pregnancy. Cutaneous necrosis with reduced activity of protein C
sometimes occurs during the first weeks of therapy. Rarely, the same process causes
frank infarction of the breast, fatty tissues, intestine, and extremities. The
patho-logic lesion associated with the hemorrhagic infarction is venous
thrombosis, suggesting that it is caused by warfarin-induced depression of
protein C synthesis.
Administration & Dosage
Treatment
with warfarin should be initiated with standard doses of 5–10 mg rather than
the large loading doses formerly used. The initial adjustment of the
prothrombin time takes about 1 week, which usually results in a maintenance
dose of 5–7 mg/d. The prothrombin time
(PT) should be increased to a level represent-ing a reduction of
prothrombin activity to 25% of normal and maintained there for long-term
therapy. When the activity is less than 20%, the warfarin dosage should be
reduced or omitted until the activity rises above 20%.
The
therapeutic range for oral anticoagulant therapy is defined in terms of an
international normalized ratio (INR). The INR is the prothrombin time ratio
(patient prothrombin time/mean of normal prothrombin time for lab)ISI,
where the ISI exponent refers to the International Sensitivity Index, and is
dependent on the specific reagents and instruments used for the determination.
The ISI serves to relate measured prothrombin times to a World Health
Organization reference standard thromboplastin; thus the prothrombin times
performed on different properly calibrated instruments with a variety of
thromboplastin reagents should give the same INR results for a given sample.
For most reagent and instrument combinations in current use, the ISI is close
to 1, mak-ing the INR roughly the ratio of the patient prothrombin time to the
mean normal prothrombin time. The recommended INR for prophylaxis and treatment
of thrombotic disease is 2–3. Patients with some types of artificial heart
valves (eg, tilting disk) or other medical conditions increasing thrombotic
risk have a recom-mended range of 2.5–3.5.
Occasionally
patients exhibit warfarin resistance, defined as progression or recurrence of a
thrombotic event while in the therapeutic range. These individuals may have
their INR target raised (which is accompanied by an increase in bleeding risk)
or be changed to an alternative form of anticoagulation (eg, daily injec-tions
of LMWH). Warfarin resistance is most commonly seen in patients with advanced
cancers, typically of gastrointestinal origin (Trousseau’s syndrome). A recent
study has demonstrated the superiority of LMWH over warfarin in preventing
recurrent venous thromboembolism in patients with cancer.
Drug Interactions
The
oral anticoagulants often interact with other drugs and with disease states.
These interactions can be broadly divided into pharmacokinetic and
pharmacodynamic effects (Table 34–2). Pharmacokinetic mechanisms for drug
interaction with oral anticoagulants are mainly enzyme induction, enzyme
inhibition, and reduced plasma protein binding. Pharmacodynamic mecha-nisms for
interactions with warfarin are synergism (impaired hemostasis, reduced clotting
factor synthesis, as in hepatic disease), competitive antagonism (vitamin K),
and an altered physiologic control loop for vitamin K (hereditary resistance to
oral anticoagulants).
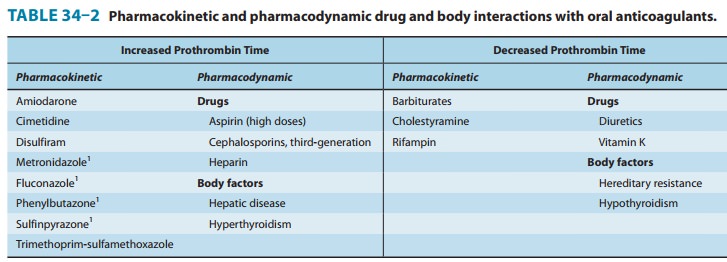
The most serious interactions with warfarin are those that increase the anticoagulant effect and the risk of bleeding. The most dangerous of these interactions are the pharmacokinetic interactions with the mostly obsolete pyrazolones phenylbuta-zone and sulfinpyrazone. These drugs not only augment the hypoprothrombinemia but also inhibit platelet function and may induce peptic ulcer disease . The mecha-nisms for their hypoprothrombinemic interaction are a stereose-lective inhibition of oxidative metabolic transformation of S-warfarin (the more potent isomer) and displacement of albu-min-bound warfarin, increasing the free fraction. For this and other reasons, neither phenylbutazone nor sulfinpyrazone is in common use in the USA. Metronidazole, fluconazole, and trimethoprim-sulfamethoxazole also stereoselectively inhibit the metabolic transformation of S-warfarin, whereas amiodarone, disulfiram, and cimetidine inhibit metabolism of both enantiomorphs f warfarin. Aspirin, hepatic disease, and hyper-thyroidism augment warfarin’s effects—aspirin by its effect on platelet function and the latter two by increasing the turnover rate of clotting factors. The third-generation cephalosporins eliminate the bacteria in the intestinal tract that produce vitamin K and, like warfarin, also directly inhibit vitamin K epoxide reductase.
Barbiturates
and rifampin cause a marked decrease
of the anti-coagulant effect by induction of the hepatic enzymes that
trans-form racemic warfarin. Cholestyramine binds warfarin in the intestine and
reduces its absorption and bioavailability.
Pharmacodynamic
reductions of anticoagulant effect occur with vitamin K (increased synthesis of
clotting factors), the diuretics chlorthalidone and spironolactone (clotting
factor concentration), hereditary resistance (mutation of vitamin K
reactivation cycle molecules), and hypothyroidism (decreased turnover rate of
clot-ting factors).
Drugs
with no significant effect on
anticoagulant therapy include ethanol, phenothiazines, benzodiazepines,
acetamino-phen, opioids, indomethacin, and most antibiotics.
Reversal of Warfarin Action
Excessive
anticoagulant effect and bleeding from warfarin can be reversed by stopping the
drug and administering oral or parenteral vitamin K1 (phytonadione),
fresh-frozen plasma, prothrombin complex concentrates such as Bebulin and
Proplex T, and recom-binant factor VIIa (rFVIIa). The disappearance of
excessive effect is not correlated with plasma warfarin concentrations but
rather with re-establishment of normal activity of the clotting factors. A
modest excess of anticoagulant effect without bleeding may require no more than
cessation of the drug. The warfarin effect can be rapidly reversed in the
setting of severe bleeding with the administration of prothrombin complex or
rFVIIa coupled with intravenous vitamin K. It is important to note that due to
the long half-life of warfarin, a single dose of vitamin K or rFVIIa may not be
sufficient.
Related Topics