Chapter: Basic & Clinical Pharmacology : Drugs Used in Disorders of Coagulation
Blood Coagulation Cascade
BLOOD COAGULATION CASCADE
Blood coagulates due
to the transformation of soluble fibrinogen into insoluble fibrin by the enzyme
thrombin. Several circulating proteins interact in a cascading series of
limited proteolytic reac-tions (Figure 34–2). At each step, a clotting factor
zymogen undergoes limited proteolysis and becomes an active protease (eg,
factor VII is converted to factor VIIa). Each protease factor activates the
next clotting factor in the sequence, culminating inthe formation of thrombin
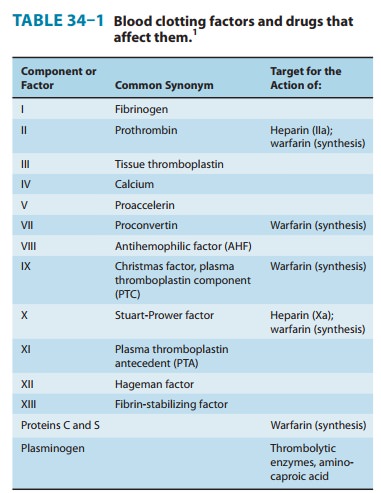
(factor IIa). Several of these factors are targets for drug therapy (Table
34–1).
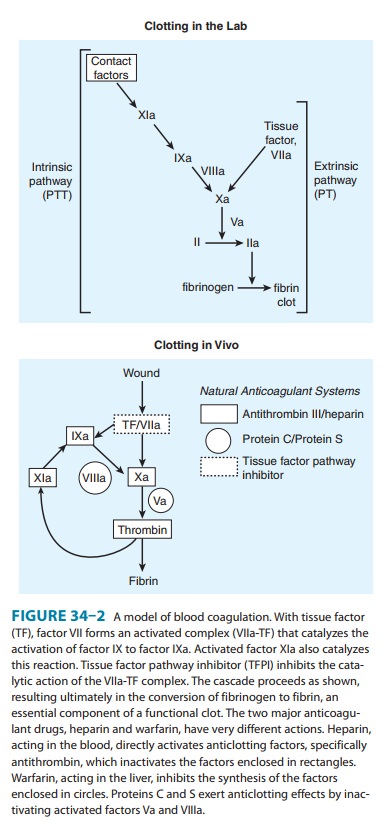
Thrombin has a central
role in hemostasis and has many func-tions. In clotting, thrombin
proteolytically cleaves small peptides from fibrinogen, allowing fibrinogen to
polymerize and form a fibrin clot. Thrombin also activates many upstream
clotting fac-tors, leading to more thrombin generation, and activates factor XIII,
a transaminase that cross-links the fibrin polymer and stabi-lizes the clot.
Thrombin is a potent platelet activator and mitogen. Thrombin also exerts anticoagulant effects by activating the
protein C pathway, which attenuates the clotting response (Figure 34–2). It
should therefore be apparent that the response to vascular injury is a complex
and precisely modulated process that ensures that under normal circumstances,
repair of vascular injury occurs with-out thrombosis and downstream ischemia;
that is, the response is proportionate and reversible. Eventually vascular
remodeling and repair occur with reversion to the quiescent resting
anticoagulant endothelial cell phenotype.
Initiation of Clotting: The Tissue Factor-VIIa Complex
The main initiator of blood coagulation in vivo is the tissue factor (TF)-factor VIIa pathway (Figure 34–2). Tissue factor is a trans-membrane protein ubiquitously expressed outside the vasculature, but not normally expressed in an active form within vessels.
The exposure of TF on damaged endothelium or to blood that has extravasated into tissue binds TF to factor VIIa. This complex, in turn, activates factors X and IX. Factor Xa along with factor Va forms the prothrombinase complex on activated cell surfaces, which catalyzes the conversion of prothrombin (factor II) to thrombin (factor IIa). Thrombin, in turn, activates upstream clotting factors, primarily factors V, VIII, and XI, resulting in amplification of thrombin generation. The TF-factor VIIa-catalyzed activation of factor Xa is regulated by tissue factor pathway inhibitor (TFPI).
Thus after initial activation of factor X to Xa by TF-VIIa, further
propagation of the clot is by feedback amplifica-tion of thrombin through the
intrinsic pathway factors VIII and IX (this provides an explanation of why
patients with deficiency of factor VIII or IX—hemophilia A and hemophilia B,
respectively— have a severe bleeding disorder).
It
is also important to note that the coagulation mechanism in vivo does not occur
in solution, but is localized to activated cellsurfaces
expressing anionic phospholipids such as phosphatidylser-ine, and is
mediated by Ca2+ bridging between the anionic
phos-pholipids and γ-carboxyglutamic
acid residues of the clotting factors. This is the basis for using calcium
chelators such as ethyl-enediamine tetraacetic acid (EDTA) or citrate to
prevent blood from clotting in a test tube.
Antithrombin (AT) is
an endogenous anticoagulant and amember of the serine protease inhibitor
(serpin) family; it inacti-vates the serine proteases IIa, IXa, Xa, XIa, and
XIIa. The endog-enous anticoagulants protein
C and protein S attenuate the
blood clotting cascade by proteolysis of the two cofactors Va and VIIIa. From
an evolutionary standpoint, it is of interest that factors V and VIII have an
identical overall domain structure and considerable homology, consistent with a
common ancestor gene; likewise the serine proteases are descendants of a trypsin-like
common ancestor. Thus, the TF–VIIa initiating complex, serine proteases, and
cofac-tors each have their own lineage-specific attenuation mechanism (Figure
34–2). Defects in natural anticoagulants result in an increased risk of venous
thrombosis. The most common defect in the natural anticoagulant system is a
mutation in factor V (factor V Leiden), which results in resistance to
inactivation by the protein C, protein S mechanism.
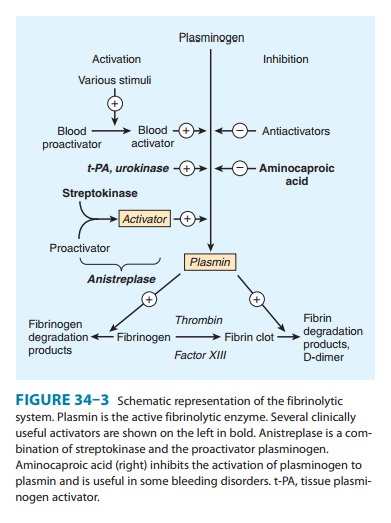
Fibrinolysis
Fibrinolysis refers to
the process of fibrin digestion by the fibrin-specific protease, plasmin. The
fibrinolytic system is similar to the coagulation system in that the precursor
form of the serine pro-tease plasmin circulates in an inactive form as
plasminogen. In response to injury, endothelial cells synthesize and release
tissue plasminogen activator (t-PA), which converts plasminogen to plasmin
(Figure 34–3). Plasmin remodels the thrombus and limits its extension by proteolytic
digestion of fibrin.
Both plasminogen and
plasmin have specialized protein domains (kringles) that bind to exposed
lysines on the fibrin clot and impart clot specificity to the fibrinolytic
process. It should be noted that this clot specificity is only observed at physiologic levels of t-PA. At the pharmacologic levels of t-PA used in
thrombolytic therapy, clot specificity is lost and a systemic lytic state is
created, with attendant increase in bleeding risk. As in the coagulation
cascade, there are negative regulators of fibrinolysis: endothelial cells
synthesize and release plasminogen activator inhibitor (PAI), which inhibits
t-PA; in addition α2 antiplasmin circulates in the blood at high concentrations and
under physiologic conditions will rapidly inactivate any plasmin that is not
clot-bound. However, this regulatory system is overwhelmed by therapeutic doses
of plasminogen activators.
If the coagulation and
fibrinolytic systems are pathologically activated, the hemostatic system may
careen out of control, lead-ing to generalized intravascular clotting and
bleeding. This process is called disseminated
intravascular coagulation (DIC) and may follow massive tissue injury,
advanced cancers, obstetric emergen-cies such as abruptio placentae or retained
products of conception, or bacterial sepsis. The treatment of DIC is to control
the underly-ing disease process; if this is not possible, DIC is often fatal.
Regulation of the
fibrinolytic system is useful in therapeutics. Increased fibrinolysis is
effective therapy for thrombotic disease. Tissue
plasminogen activator, urokinase, and
streptokinase allactivate the fibrinolytic system (Figure 34–3).
Conversely, decreased fibrinolysis protects clots from lysis and reduces the
bleeding of hemostatic failure. Aminocaproic
acid is a clinically useful inhibitor of fibrinolysis. Heparin and the oral
anticoagulant drugs do not affect the fibrinolytic mechanism.
Related Topics