Chapter: Basic & Clinical Pharmacology : Drugs Used in Disorders of Coagulation
Drugs Used in Bleeding Disorders
DRUGS USED IN BLEEDING DISORDERS
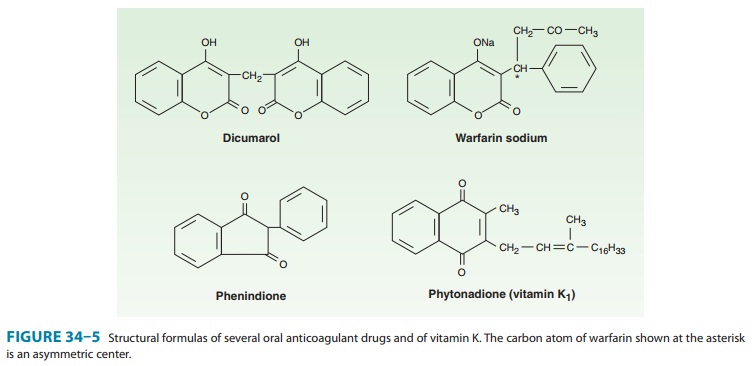
VITAMIN K
Vitamin K confers biologic
activity upon prothrombin and factors VII, IX, and X by participating in their
postribosomal modifica-tion. Vitamin K is a fat-soluble substance found
primarily in leafy green vegetables. The dietary requirement is low, because
the vita-min is additionally synthesized by bacteria that colonize the human
intestine. Two natural forms exist: vitamins K1 and K2.
Vitamin K1 (phytonadione; Figure 34–5) is found in food. Vitamin K2
(menaquinone) is found in human tissues and is syn-thesized by intestinal bacteria.Vitamins
K1 and K2 require bile salts for absorption from the
intestinal tract. Vitamin K1 is available clinically in oral and
par-enteral forms. Onset of effect is delayed for 6 hours but the effect is
complete by 24 hours when treating depression of prothrombin activity by excess
warfarin or vitamin K deficiency. Intravenous administration of vitamin K1
should be slow, because rapid infu-sion can produce dyspnea, chest and back
pain, and even death. Vitamin K repletion is best achieved with intravenous or
oral administration, because its bioavailability after subcutaneous
administration is erratic. Vitamin K1 is currently administered to
all newborns to prevent the hemorrhagic disease of vitamin K deficiency, which
is especially common in premature infants. The
water-soluble salt of vitamin K3 (menadione) should never be used in therapeutics. It is
particularly ineffective in the treatment of war-farin overdosage. Vitamin K
deficiency frequently occurs in hospi-talized patients in intensive care units
because of poor diet, parenteral nutrition, recent surgery, multiple antibiotic
therapy, and uremia. Severe hepatic failure results in diminished protein
synthe-sis and a hemorrhagic diathesis that is unresponsive to vitamin K.
PLASMA FRACTIONS
Sources & Preparations
Deficiencies
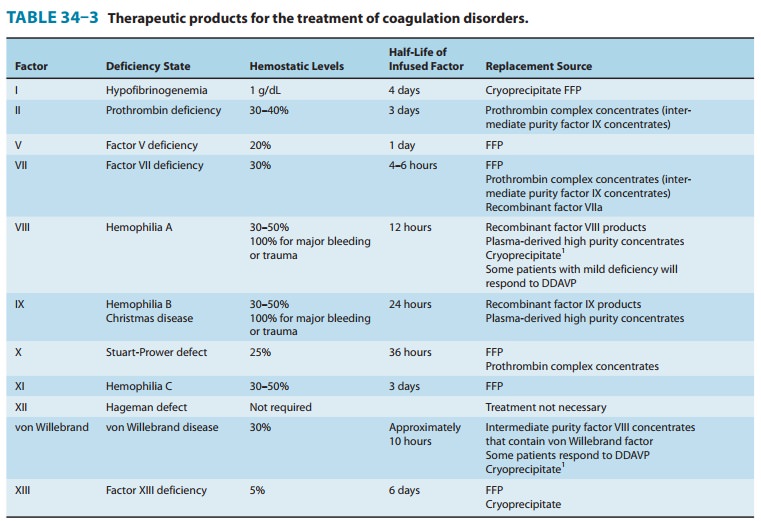
in plasma coagulation factors can cause bleeding (Table 34–3). Spontaneous
bleeding occurs when factor activity is less than 5–10% of normal. Factor VIII
deficiency (classichemophilia, or hemophilia A) and factor IX deficiency (Christmas disease, or hemophilia B) account for most of
theheritable coagulation defects. Concentrated plasma fractions are available
for the treatment of these deficiencies. Administration of plasma-derived,
heat- or detergent-treated factor concen-trates and recombinant factor
concentrates are the standard treatments for bleeding associated with
hemophilia. Lyophilized factor VIII concentrates are prepared from large pools
of plasma. Transmission of viral diseases such as hepatitis B and C and HIV is
reduced or eliminated by pasteurization and by extraction of plasma with
solvents and detergents. However, this treatment does not remove other
potential causes of transmissible diseases such as prions. For this reason,
recombinant clotting factor preparations are recommended whenever possible for
factor replacement. The best use of these therapeutic materials requires
diagnostic specificity of the deficient factor and quantitation of its activity
in plasma. Intermediate purity factor VIII concen-trates (as opposed to
recombinant or high purity concentrates) contain significant amounts of von
Willebrand factor. Humate-P is a factor VIII concentrate that is approved by
the FDA for the treatment of bleeding associated with von Willebrand disease.
Clinical Uses
An uncomplicated hemorrhage into a joint should be treated with sufficient factor VIII or factor IX replacement to maintain a level of at least 30–50% of the normal concentration for 24 hours. Soft tissue hematomas require a minimum of 100% activity for 7 days. Hematuria requires at least 10% activity for 3 days. Surgery and major trauma require a minimum of 100% activity for 10 days.
The initial loading dose
for factor VIII is 50 units/kg of body weight to achieve 100% activity of
factor VIII from a baseline of 1% or less, assuming a normal hemoglobin. Each
unit of factor VIII per kilogram of body weight raises its activity in plasma
2%. Replacement should be administered every 12 hours. Factor IX therapy
requires twice the dose of factor VIII, but with an admin-istration of about
every 24 hours because of its longer half-life. Recombinant factor IX has only
80% recovery compared with plasma-derived factor IX products. Therefore, dosing
with recom-binant factor IX requires 120% of the dose used with the
plasma-derived product.
Desmopressin acetate increases
the factor VIII activity ofpatients with mild hemophilia A or von Willebrand
disease. It can be used in preparation for minor surgery such as tooth
extraction without any requirement for infusion of clotting factors if the
patient has a documented adequate response. High-dose intrana-sal
desmopressin is available and has been
shown to be efficacious and well tolerated by patients.
Freeze-dried
concentrates of plasma containing prothrombin, factors IX and X, and varied
amounts of factor VII (Proplex, etc) are commercially available for treating
deficiencies of these factors (Table 34–3). Each unit of factor IX per kilogram
of body weight raises its activity in plasma 1.5%. Heparin is often added to
inhibit coagulation factors activated by the manufacturing pro-cess. However,
addition of heparin does not eliminate all throm-boembolic risk.
Some
preparations of factor IX concentrate contain activated clotting factors, which has led to their use in treating
patients with inhibitors or antibodies to factor VIII or factor IX. Two
products are available expressly for this purpose: Autoplex (with factor VIII correctional activity) and FEIBA (Factor Eight Inhibitor Bypassing Activity).
These products are not uniformlysuccessful in arresting hemorrhage, and the
factor IX inhibitor titers often rise after treatment with them. Acquired
inhibitors of coagulation factors may also be treated with porcine factor VIII
(for factor VIII inhibitors) and recombinant activated factor VII. Recombinant
activated factor VII (NovoSeven) is
being increas-ingly used to treat coagulopathy associated with liver disease
and major blood loss in trauma and surgery. These recombinant and
plasma-derived factor concentrates are very expensive, and the indications for
them are very precise. Therefore, close consultation with a hematologist
knowledgeable in this area is essential.
Cryoprecipitate is
a plasma protein fraction obtainable fromwhole blood. It is used to treat
deficiencies or qualitative abnor-malities of fibrinogen, such as that which
occurs with dissemi-nated intravascular coagulation and liver disease. A single
unit of cryoprecipitate contains 300 mg of fibrinogen.
Cryoprecipitate
may also be used for patients with factor VIII deficiency and von Willebrand
disease if desmopressin is not indi-cated and a pathogen-inactivated,
recombinant, or plasma-derived product is not available. The concentration of
factor VIII and von Willebrand factor in cryoprecipitate is not as great as
that found in the concentrated plasma fractions. Moreover, cryoprecipitate is
not treated in any manner to decrease the risk of viral exposure. For infusion,
the frozen cryoprecipitate unit is thawed and dissolved in a small volume of
sterile citrate-saline solution and pooled with other units. Rh-negative women
with potential for childbearing should receive only Rh-negative cryoprecipitate
because of possible contamination of the product with Rh-positive blood cells.
RECOMBINANT FACTOR VIIA
Recombinant factor
VIIa is approved for treatment of inherited or acquired hemophilia A or B with
inhibitors, treatment of bleeding associated with invasive procedures in
congenital or acquired hemophilia, or factor VII deficiency. In the EU, the
drug is also approved for treatment of Glanzmann’s thrombasthenia.
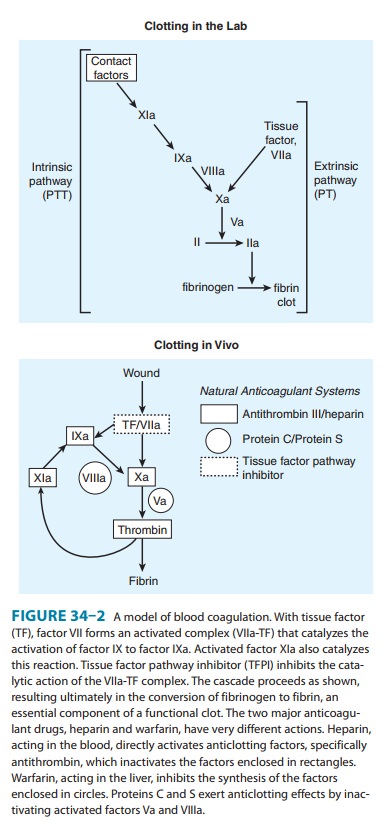
Factor
VIIa initiates activation of the clotting pathway by acti-vating factor IX and
factor X in association with tissue factor (see Figure 34–2). The drug is given
by bolus injection. For hemo-philia A or B with inhibitors and bleeding, the
dose is 90 mg/kg every 2 hours until hemostasis is achieved, and then continued
at 3–6 hour intervals until stable. For congenital factor VII defi-ciency, the
recommended dosage is 15–30 mg/kg every 4–6 hours until hemostasis is achieved.
Factor
VIIa has been widely used for off-label indications, including bleeding with
trauma, surgery, intracerebral hemorrhage, and warfarin toxicity. A major
concern of off-label use has been the possibility that thrombotic events may be
increased. A recent study examined rates of thromboembolic events in 35
placebo-controlled trials where factor VIIa was administered for nonapproved
indica-tions. This study found an increase in arterial, but not venous,
thrombotic events, particularly among elderly individuals.
FIBRINOLYTIC INHIBITORS: AMINOCAPROIC ACID
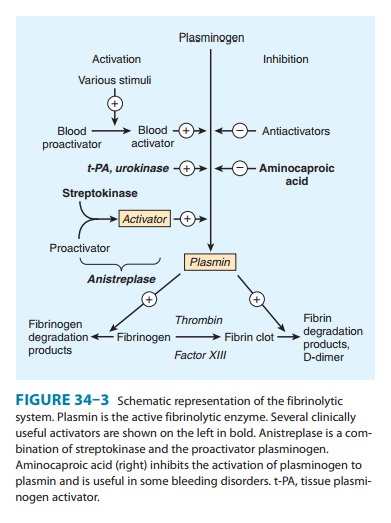
Aminocaproic
acid (EACA), which is chemically similar to the amino acid lysine, is a
synthetic inhibitor of fibrinolysis. It com-petitively inhibits plasminogen activation
(Figure 34–3). It is rap-idly absorbed orally and is cleared from the body by
the kidney. The usual oral dosage of EACA is 6 g four times a day. When the
drug is administered intravenously, a 5 g loading dose should be infused over
30 minutes to avoid hypotension. Tranexamic
acid is an ana-log of aminocaproic acid and has the same properties. It is
adminis-tered orally with a 15 mg/kg loading dose followed by 30 mg/kg every 6
hours.
Clinical
uses of EACA are as adjunctive therapy in hemophilia, as therapy for bleeding
from fibrinolytic therapy, and as prophy-laxis for rebleeding from intracranial
aneurysms. Treatment success has also been reported in patients with
postsurgical gastrointestinal bleeding and postprostatectomy bleeding and
bladder hemorrhage secondary to radiation- and drug-induced cystitis. Adverse
effects of the drug include intravascular thrombosis from inhibition of
plasminogen activator, hypotension, myopathy, abdominal dis-comfort, diarrhea,
and nasal stuffiness. The drug should not be used in patients with disseminated
intravascular coagulation or genitourinary bleeding of the upper tract, eg,
kidney and ureters, because of the potential for excessive clotting.
SERINE PROTEASE INHIBITORS: APROTININ
Aprotinin
is a serine protease inhibitor (serpin) that inhibits fibrinolysis by free
plasmin and may have other antihemorrhagic effects as well. It also inhibits
the plasmin-streptokinase complex in patients who have received that
thrombolytic agent. Aprotinin was shown to reduce bleeding—by as much as
50%—from many types of surgery, especially that involving extracorporeal
circula-tion for open heart procedures and liver transplantation. However,
clinical trials and internal data from the manufacturer suggested that use of
the drug was associated with an increased risk of renal failure, heart attack,
and stroke. A prospective trial was initiated in Canada but halted early
because of concerns that use of the drug was associated with increased
mortality. The drug was removed from the market in 2007.
Related Topics