Chapter: Genetics and Molecular Biology: Protein Structure
Thermodynamic Considerations of Protein Structure
Thermodynamic Considerations of Protein
Structure
Thermodynamics
provides a useful framework for calculation of equilibrium constants of
reactions. This also applies to the “reaction” of protein denaturation.
Consider a protein denaturing from a specific native conformation, N, to any of a great many nonspecific,
random conformations characteristic of denatured proteins, D. The reaction can be described by an equilibrium constant that
relates the amount of the protein found in each of the two states if the system
has reached equilibrium,
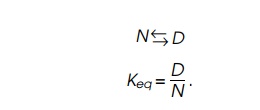
Thermodynamics
provides a way of calculating Keq
as
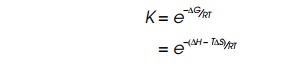
where ∆G is the
change in Gibbs free energy; R is the
universal gas constant; T is the
absolute temperature in degrees Kelvin; ∆H is the
enthalpy change of the reaction, which in biological systems is equiva-lent to
binding energy when volume changes can be neglected; and ∆S is the
entropy change of the reaction. Entropy is related to the number of equivalent
states of a system. The state of a protein molecule confined to one
conformation without any degrees of freedom possesses much lower entropy than a
denatured protein that can adopt any of a great number of conformations all at
the same energy. For clarity, we will neglect the contributions of the
surrounding water in further considera-tions, but in physically meaningful
calculations these too must be included.
Let us
examine why proteins denature when the temperature is raised. If the protein is
in the folded state at the lower temperature, Keq is less than 1, that is,
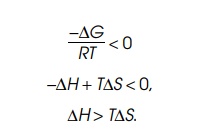
∆H>T∆S.
As the
temperature increases, neglecting the small temperature-dependent changes that
occur in the interaction energies and entropy change, the term T∆S
increases, and eventually exceeds ∆H. Then
the equilibrium shifts to favor the denatured state.
The
temperature dependence of the denaturing of proteins provides the information
necessary for determination of ∆Η of
denaturing. It is very large! This means that ∆S for
denaturing is also very large, just as we inferred above, and at temperatures
near the denaturing point, the
difference
of these two large numbers barely favors retention of the structure of the
protein. Hence the binding energies of the many interactions that determine
protein structure, hydrogen bonds, salt bridges, dipole-dipole interactions,
dispersion forces, and hydrophobic forces just barely overcome the disruptive
forces. Thus we see the value of the peptide bond. If rotations about the C-N
bond were not re-stricted,the increased degrees of freedom available to the
protein would be enormous. Then the energy and entropy balance would be tipped
in the direction of denatured proteins.
Related Topics