Chapter: Genetics and Molecular Biology: Protein Structure
Secondary Structure Predictions - Protein Structure
Secondary Structure Predictions
Less ambitious than calculating the tertiary
structure of a protein is predicting its secondary structure. There is some
hope that this is a much simpler problem than prediction of tertiary structure
because most of the interactions determining secondary structure at an amino
acid residue derive from amino acids close by in the primary sequence. The
problem is how many amino acids need to be considered and how likely is a
prediction to be correct? We can estimate both by utilizing information from
proteins whose structures are known. The question is how long a stretch of
amino acids is required to specify a secondary structure. For example, if a
stretch of five amino acids were sufficient, then the same sequence of five
amino acids ought to adopt the same structure, regardless of the protein in
which they occur.
The tertiary structures available from X-ray
diffraction studies can be used as input data. Many examples now exist where a sequence
of five amino acids appears in more than one protein. In about 60% of these
cases, the same sequence of five amino acids is found in the same secondary
structure. Of course, not all possible five amino acid se-quences are
represented in the sample set, but the set is sufficiently large that it is
clear that we should not expect to have better than about 60% accuracy in any
secondary structure prediction scheme if we consider only five amino acids at a
time.
Several approaches have been used to determine
secondary structure prediction rules. At one end is a scheme based on the known
conforma-tions assumed by homopolymers and extended by analysis of a small
number of known protein structures. The Chou and Fasman approach is in this
category. A more general approach is to use information theory to generate a
defined algorithm for predicting secondary structure. This overcomes many of
the ambiguities of the Chou-Fasman prediction scheme.
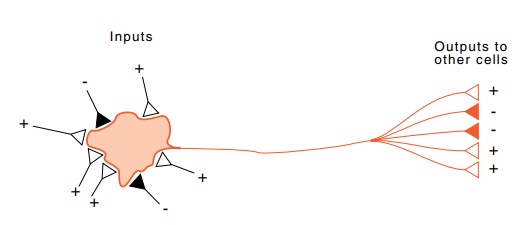
Recently, neural networks have been applied to
predicting secondary structure. While usually implemented on ordinary
computers, these simulate on a crude scale some of the known properties of
neural connections in parts of the brain. Depending on the sum of the positive
and negative inputs, a neuron either does not fire, or fires and sends
activating and inhibiting signals on to the neurons its output is con-nected to
In predicting secondary structure by a neural
network, the input is the identity of each amino acid in a stretch of ten to
fifteen amino acids (Fig. 6.17). Since each of these can be any of the twenty
amino acids, about 200 input lines or “neurons” are on this layer. Each of
these activates or inhibits each neuron on a second layer by a strength that is
adjusted by training. After summing the positive and negative signals reaching
it, a neuron on the second layer either tends to “fire” and sends a strong
activating or inhibiting signal on to the third layer, or it tends not to fire.
In the case of protein structure prediction, there would be three neurons in
the third layer. One corresponds to predicting α-helix, one to β-sheet,
and one to random coil. For a given input sequence, the network’s secondary
structure prediction for the central amino acid of
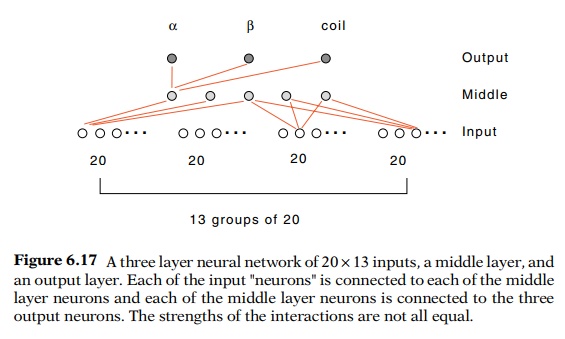
A three
layer neural network of 20 × 13
inputs, a middle layer, and an output layer. Each of the input
"neurons" is connected to each of the middle layer neurons and each
of the middle layer neurons is connected to the three output neurons. The
strengths of the interactions are not all equal.
the
sequence is considered to correspond to the neuron of the third layer with the
highest output value. “Training” such a network is done by presenting various
stretches of amino acids whose secondary structure is known and adjusting the
strengths of the interactions between neu-rons so that the network predicts the
structures correctly.
No matter
what scheme is used, the accuracy of the resulting struc-ture prediction rules
never exceeds about 65%. Note that a scheme with no predictive powers
whatsoever would be correct for about 33% of the amino acids in a protein. The
failure of these approaches to do better than 65% means that in some cases,
longer-range interactions between amino
acids in a protein have a significant effect in determining secon-dary
structure (See problem 6.18).
Related Topics