Chapter: Genetics and Molecular Biology: Transcription,Termination, and RNA Processing
The Discovery and Assay of RNA Splicing
The Discovery and Assay of RNA Splicing
The discovery by Berget and Sharp and by Roberts and co-workers that segments within newly synthesized messenger RNA could be eliminated before the RNA was exported from the nucleus to the cytoplasm was a great surprise. Not only was the enzymology of such a reaction unfamiliar, but the biological need for such a reaction was not apparent. Although splicing is a possible point for cells to regulate expression, there seemed no particular need for utilizing this possibility. One possible reason for splicing could be in the evolution of proteins. Intervening sequences place genetic spacers between coding regions in a gene. As a result, recombination is more likely to occur between coding regions than within them. This permits a coding region to be inherited as an independent module. Not surprisingly then, the portions of pro-teins that such modules encode often are localized structural domains within proteins. As a result, during the evolution of a protein, domains may be shuffled. It is hard to imagine, however, that this need was great enough to drive the evolution of splicing.
The most plausible explanation for the existence of
intervening se-quences is that they are the remnants of a parasitic sequence
that spread through the genome of some early cell-type. In order that the
sequence not inactivate a coding region into which it inserted itself, the
coding region arranged that it splice itself out of mRNA. Thus, although the
parasitic sequence might have inserted into the middle of an essential gene,
the gene was not inactivated. After transcription, and before translation, the
RNA copy of the gene containing the sequence was cut and spliced to recover the
intact uninterrupted gene. Now that the intervening sequences are there, the
cell is beginning to make use of them. One example is regulating the use of
alternative splice sites to generate one or another gene product from a single
gene.
Both the discovery of messenger splicing and a
clear demonstration of splicing used adenovirus RNA. The virus provided a
convenient source of DNA for hybridization reactions and RNA extracted from
adeno-infected cultured cells was a rich source of viral RNA. Electron
microscopy of hybrids formed between adenovirus mRNA and a frag-ment of the
adenovirus genome coding for the coat hexon protein was performed to locate the
transcriptional unit. Curiously, the hybrids which formed lacked the expected
structure. Many nucleotides at one
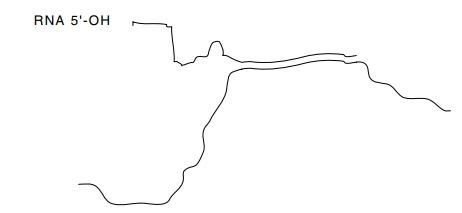
Figure
5.13 RNA-DNA hybrid between a
single-stranded fragment of ade-novirus DNA and hexon mRNA extracted from
cells. The RNA and DNA are not complementary at the 5’ end of the mRNA.
Figure
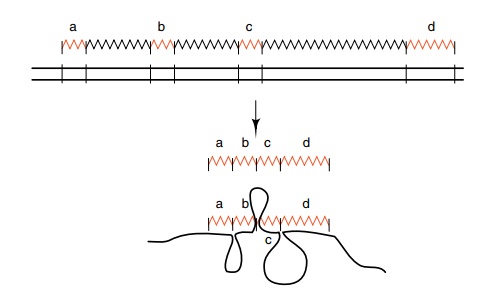
5.14 Hybridization of adenovirus hexon
mRNA to adenovirus DNAfrom a region well upstream from the hexon gene. The
regions a, b, c and d of viral RNA hybridize, but the regions between these
segments are missing from the RNA, and the DNA accordingly loops out.
end of the RNA failed to hybridize to the hexon DNA
(Fig. 5.13). One of the ways splicing was discovered was tracking down the
source of this extraneous RNA. Further experiments with longer DNA fragments
located DNA complementary to this RNA, but it was far away from the virus hexon
gene (Fig. 5.14). The RNA as extracted from the cells had to have been
synthesized, and then cut and spliced several times.
Gel electrophoresis provides a convenient assay to
detect splicing and to locate the spliced regions. Hybridization of DNA to
messenger RNA
Figure
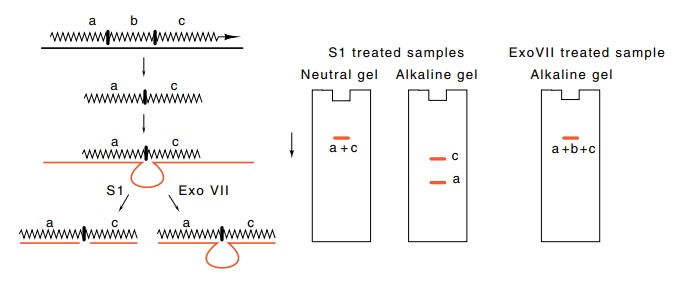
5.15 Endonuclease S1 is used to digest
single-stranded regions of RNAand DNA, and exonuclease Exo VII is used to
digest single-stranded DNA exonucleolytically. The DNA is labeled in these
experiments. After digestions, electrophoresis on neutral gels analyzes
duplexes, and electrophoresis on alka-line gels denatures and analyzes the
sizes of fragments.
that has been spliced to remove introns yields
duplexes and loops as shown in Fig. 5.15. The locations and sizes can be
determined by digestion with S1 and ExoVII nucleases followed by
electrophoresis on denaturing gels and on gels that preserve the integrity of
RNA-DNA duplexes. Since S1 nuclease digests single-stranded RNA and DNA and can
act endonucleolytically, it removes all single-stranded or unpaired regions
from RNA and DNA. ExoVII, however, digests only single-stranded DNA, and only from
the ends. This enzyme therefore provides the additional information necessary
to determine intron and exon sizes and locations. After digestion with these
enzymes, the oligonucleotides can be separated according to size by
electrophoresis on polyacrylamide gels. Gels run with normal buffer near
neutral pH provide the sizes of the double-stranded duplex molecules, and gels
run at alkaline pH provide the molecular weights of the denatured,
single-stranded oli-gonucleotides.
Related Topics