Chapter: Medicine and surgery: Genitourinary system
Renal tubular acidosis - Tubular and interstitial diseases
Renal tubular acidosis
Definition
These are syndromes in which a metabolic disorder of tubular function is the main feature. They may be inherited or acquired.
Aetiology
They may be classified as single or multiple defects, or they can be grouped according to the part of the nephron affected (see Fig. 6.9). See also Renal Tubular Acidosis (see below). The single defects are discussed here.
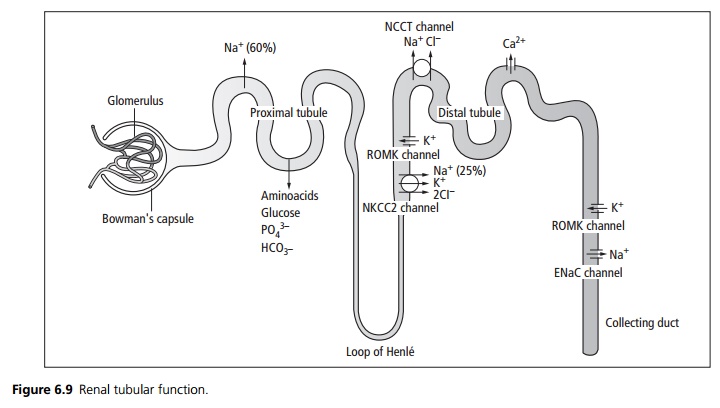
Proximal tubule: The proximal tubule is the site of maximal reabsorption of glucose and sodium. Water and anions such as aminoacids follow sodium. Osmotic diuretics and carbonic anhydrase inhibitors act at this site. Disorders of the proximal tubule may lead to one or more of the following syndromes:
┬Ę Renal glycosuria is an autosomal recessive inherited condition characterised by glycosuria with normal blood glucose. There is impaired reabsorption of glucose in the proximal tubules, with no clinical sequelae. Glycosuria is a normal response during pregnancy.
┬Ę Aminoaciduria may affect only one amino acid or several. The most important single defect is cystinuria, an autosomal recessive condition which predisposes to urinary stone formation. Treatment is with high fluid intake and alkali ingestion, because the cystine is more soluble in alkaline conditions. Drugs such as penicillamine may be used to form complexes with the cystine. In Fanconi syndrome (which may be an inherited juvenile form or acquired in adults) there is defective reabsorption of most aminoacids, glucose, phosphate and bicarbonate. There may be potassium depletion, polyuria and immunodeficiency secondary to immunoglobulin loss. Phosphate loss leads to vitamin D resistant rickets in childhood, and osteomalacia in adults.
┬Ę Phosphate transport defects: There are several types, usually X-linked, although occasional sporadic inherited or acquired cases do occur. They cause inappropriate loss of phosphate from the tubules and result in hypophosphataemia and vitamin D resistant rickets (VDDR). Treatment is with oral phosphate supplements with vitamin D or 1,25 dihydroxyvitamin D
┬Ę (calcitriol).
Thick ascending loop of Henle: Sodium is pumped out of the lumen in the ascending loop, water is drawn out of the descending loop by osmosis. This creates a concentration gradient within the medulla of the kidney, which draws water out of the collecting duct and hence concentrates the urine. Loop diuretics such as furosemide act from within the lumen of the ascending loop binding to the chloride site of the NKCC2 channel. This interferes with the pump reducing the concentration gradient resulting in more dilute urine.
┬Ę Bartter type I syndrome is an autosomal recessive defect in the gene encoding the NKCC2 pump. It results in high urinary sodium loss, dehydration, secondary hyperaldosteronism and hypokalaemic alkalosis. There is hypercalciuria and nephrocalcinosis.
┬Ę Bartter type II syndrome is an autosomal recessive defect in the gene encoding the ROMK channel. Decreased potassium excretion causes hyperkalaemia, which interferes with the action of the NKCC2 pump. This results in a similar syndrome of sodium loss, dehydration and hypercalciuria as Bartter type I; how-ever, hypokalaemia only occurs after treatment with sodium supplements.
Distal convoluted tubule: Thiazide diuretics act from within the lumen of the distal tubule binding to the chloride site of the NCCT channel reducing sodium and water reabsorption. Calcium reabsorption is increased leading to hypercalcaemia.
┬Ę Gittleman syndrome is an autosomal recessive mutation in the gene encoding the NCCT channel. As there is increased sodium in the urine reaching the collecting duct there is exchange of sodium for potassium in the collecting duct resulting in a hypokalaemic metabolic alkalosis.
Collecting duct: At the collecting duct there is reabsorption of sodium in exchange for potassium excretion. This is under the influence of aldosterone which increases sodium (and hence water) reabsorption. Spironolactone and amiloride affect this exchange and hence increase urinary water and sodium loss. In contrast to other diuretics, these cause potassium reabsorption and are termed potassiumsparing diuretics. The permeability of the collecting duct is under the influence of anti diuretic hormone (ADH or vasopressin).
┬Ę In diabetes insipidus (see also page 445) the action of ADH is impaired. This results in excessive water loss in the urine.
Renal tubular acidosis
Definition
Renal tubular acidosis (RTA) is a metabolic acidosis of renal origin, characterised by hyperchloraemic metabolic acidosis with a normal anion gap.
Aetiology/pathophysiology
RTA may be inherited or acquired. Under physiological conditions, the kidneys help to maintain acidŌĆōbase balance, together with the lungs (which remove carbon dioxide). RTA is classified into three main types each with differing pathophysiology.
Type 1 (Distal RTA) causes include primary, idiopathic and connective tissue disorders such as SjogrenŌĆÖs┬© and rheumatoid arthritis. Impaired H+secretion in the distal tubule leads to progressive accumulation of acid in the body. Even when bicarbonate levels fall to as low as 10 mmol/L or below, the urine remains relatively alkaline (pH Ōēź 5.5). If untreated, persistent metabolic acidosis leads to increased mobilisation of calcium phosphate from bone, and this causes hypercalciuria promoting the formation of renal stones. Type 1 RTA is treated with bicarbonate replacement, with correction of hyperkalaemia.
Type 2 (Proximal RTA) usually occurs in the context of a general proximal tubule disorder called Fanconi syndrome, which may be familial inherited, idiopathic or acquired, e.g. due to drugs, heavy metals or myeloma. The proximal tubule fails to reabsorb the HCO3ŌłÆ , so this passes to the distal tubules. Once plasma bicarbonate levels fall to about 12ŌĆō16 mmol/L, the distal tubule is able to reabsorb the amount of filtered bicarbonate, so severe acidosis does not occur. The main problems occur due to the loss of other substances such as amino acids and phosphate. Type 2 is treated with bicarbonate, thiazide diuretic and potassium bicarbonate or potassium-sparing diuretics. Fanconi syndrome is treated with large doses of vitamin D.
Type 4 is hypoaldosteronism or resistance to the effects of aldosterone such as caused by mild renal failure, interstitial nephritis or potassiumsparing diuretics like spironalactone. It appears that aldosterone deficiency causes hyperkalaemia, which is associated with a mild metabolic acidosis. Urinary pH usually remains appropriately low (<5.3). Mineralocorticoid deficiency is corrected using fludrocortisone. Hyper-kalaemia may be life-threatening and the underlying disorder often shortens life expectancy.
Related Topics