Chapter: Basic & Clinical Pharmacology : Drug Receptors & Pharmacodynamics
Relation Between Drug Concentration & Response
RELATION BETWEEN DRUG
CONCENTRATION & RESPONSE
The
relation between dose of a drug and the clinically observed response may be
complex. In carefully controlled in vitro systems, however, the relation
between concentration of a drug and its effect is often simple and can be
described with mathematicalprecision. This idealized relation underlies the
more complex rela-tions between dose and effect that occur when drugs are given
to patients.
Concentration-Effect Curves & Receptor Binding of Agonists
Even
in intact animals or patients, responses to low doses of a drug usually
increase in direct proportion to dose. As doses increase, however, the response
increment diminishes; finally, doses may be reached at which no further
increase in response can be achieved. In idealized or in vitro systems, the
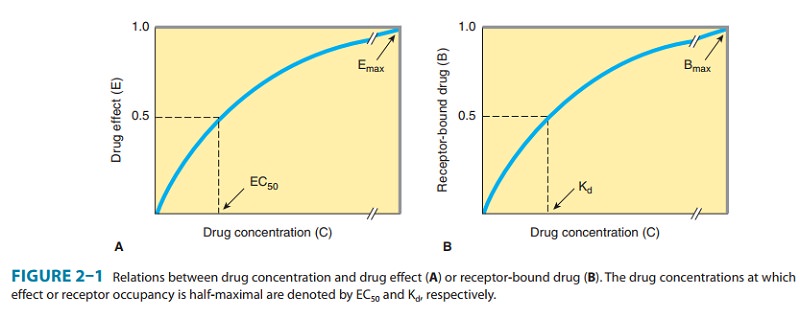
relation between drug concen-tration and effect is described by a hyperbolic
curve (Figure 2–1A) according to the following equation:
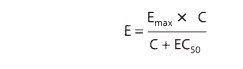
where
E is the effect observed at concentration C, Emax is the maximal
response that can be produced by the drug, and EC50 is the
concentration of drug that produces 50% of maximal effect.
This
hyperbolic relation resembles the mass action law, which describes association
between two molecules of a given affinity. This resemblance suggests that drug
agonists act by binding to (“occupy-ing”) a distinct class of biologic
molecules with a characteristic affin-ity for the drug receptor. Radioactive
receptor ligands have been used to confirm this occupancy assumption in many drug-receptor
sys-tems. In these systems, drug bound to receptors (B) relates to the
concentration of free (unbound) drug (C) as depicted in Figure 2–1B and as
described by an analogous equation:
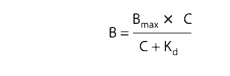
in
which Bmax indicates the total concentration of receptor sites (ie,
sites bound to the drug at infinitely high concentrations of free drug) and Kd
(the equilibrium dissociation constant) represents the concentration of free
drug at which half-maximal binding is observed. This constant characterizes the
receptor’s affinity for binding the drug in a reciprocal fashion: If the Kd
is low, binding affinity is high, and vice versa. The EC50 and Kd
may be identical, but need not be, as discussed below. Dose-response data are
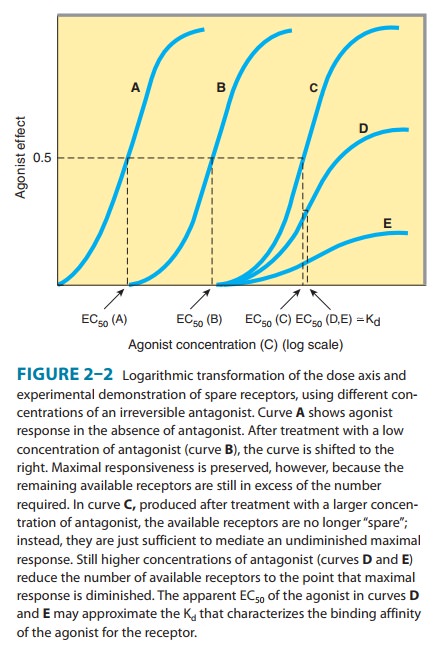
often presented as a plot of the drug effect (ordinate) against the loga-rithm of the dose or concentration
(abscissa). This mathematicalmaneuver transforms the hyperbolic curve of Figure
2–1 into a sigmoid curve with a linear midportion (eg, Figure 2–2). This
expands the scale of the concentration axis at low concentrations (where the
effect is changing rapidly) and compresses it at high concentrations (where the
effect is changing slowly), but has no special biologic or pharmacologic
significance.
Receptor-Effector Coupling & Spare Receptors
When a receptor is occupied by an agonist, the resulting
conforma-tional change is only the first of many steps usually required
toproduce a pharmacologic response. The transduction process that links drug occupancy
of receptors and pharmacologic response is often termed coupling. The relative efficiency of occupancy-response coupling is
partially determined by the initial conformational change
in
the receptor; thus, the effects of full agonists can be considered more
efficiently coupled to receptor occupancy than can the effects of partial
agonists (described in text that follows). Coupling effi-ciency is also
determined by the biochemical events that transduce receptor occupancy into
cellular response. Sometimes the biologic effect of the drug is linearly
related to the number of receptors bound. This is often true for drug-regulated
ion channels, eg, in which the ion current produced by the drug is directly
proportional to the number of receptors (ion channels) bound. In other cases,
the biologic response is a more complex function of drug binding to receptors.
This is often true for receptors linked to enzymatic signal transduction
cascades, eg, in which the biologic response often increases disproportionately
to the number of receptors occupied by drug.
Many
factors can contribute to nonlinear occupancy-response coupling, and often
these factors are only partially understood. The concept of “spare” receptors, regardless of the
precise bio-chemical mechanism involved, can help us to think about these
effects. Receptors are said to be “spare” for a given pharmacologic response if
it is possible to elicit a maximal biologic response at a concentration of
agonist that does not result in occupancy of the full complement of available
receptors. Experimentally, spare receptors may be demonstrated by using
irreversible antagonists to prevent binding of agonist to a proportion of available
receptors and showing that high concentrations of agonist can still produce an
undiminished maximal response (Figure 2–2). Thus, the same maximal inotropic
response of heart muscle to catecholamines can be elicited even under
conditions in which 90% of the β adreno-ceptors are occupied by a
quasi-irreversible antagonist. Accordingly, myocardial cells are said to
contain a large proportion of spare β adrenoceptors.
How
can we account for the phenomenon of spare receptors? In the example of the β adrenoceptor,
receptor activation pro-motes binding of guanosine triphosphate (GTP) to an
interme-diate signaling protein and activation of the signaling intermediate
may greatly outlast the agonist-receptor interaction (see the following section
on G Proteins & Second Messengers). In such a case, the “spareness” of
receptors is temporal. Maximal
response can be elicited by activation of relatively few receptors because the
response initiated by an individual ligand-receptor binding event persists
longer than the binding event itself.
In
other cases, in which the biochemical mechanism is not understood, we imagine
that the receptors might be spare in
num-ber. If the concentration or amount of cellular components otherthan
the receptors limits the coupling of receptor occupancy to response, then a
maximal response can occur without occupancy of all receptors. Thus, the
sensitivity of a cell or tissue to a particu-lar concentration of agonist
depends not only on the affinity of
the receptor for binding the agonist (characterized by the Kd) but
also on the degree of spareness—the
total number of receptors present compared with the number actually needed to
elicit a maximal biologic response.
The
concept of spare receptors is very useful clinically because it allows one to
think precisely about the effects of drug dosage without needing to consider
biochemical details of the signaling response. The Kd of the
agonist-receptor interaction determines what fraction (B/Bmax) of
total receptors will be occupied at a given free concentration (C) of agonist
regardless of the receptor concentration:
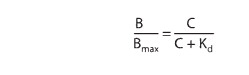
Imagine
a responding cell with four receptors and four effec-tors. Here the number of
effectors does not limit the maximal response, and the receptors are not spare in number. Consequently, an
agonist present at a concentration equal to the Kd will occupy 50%
of the receptors, and half of the effectors will be activated, producing a
half-maximal response (ie, two receptors stimulate two effectors). Now imagine
that the number of receptors increases 10-fold to 40 receptors but that the
total number of effectors remains constant. Most of the receptors are now spare
in number. As a result, a much lower concentration of agonist suf-fices to
occupy 2 of the 40 receptors (5% of the receptors), and this same low
concentration of agonist is able to elicit a half-maximal response (two of four
effectors activated). Thus, it is possible to change the sensitivity of tissues
with spare receptors by changing receptor number.
Competitive & Irreversible Antagonists
Receptor
antagonists bind to receptors but do not activate them. The primary action of
antagonists is to prevent agonists (other drugs or endogenous regulatory
molecules) from activating recep-tors. Some antagonists (so-called “inverse
agonists,”), also reduce receptor activity below basal levels observed in the
absence of bound ligand. Antagonists are divided into two classes depending on
whether or not they reversibly compete
with agonists for binding to receptors.
In
the presence of a fixed concentration of agonist, increasing concentrations of
a reversible competitive antagonist
progres-sively inhibit the agonist response; high antagonist concentra-tions
prevent response completely. Conversely, sufficiently high concentrations of
agonist can surmount the effect of a given con-centration of the antagonist;
that is, the E max for the agonist remains the same for any fixed
concentration of antagonist (Figure 2–3A). Because the antagonism is
competitive, the pres-ence of antagonist increases the agonist concentration
required
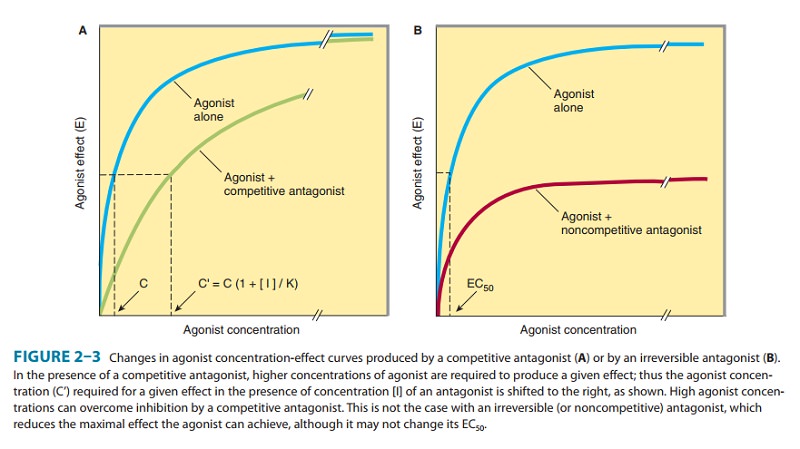
for
a given degree of response, and so the agonist concentration-effect curve is
shifted to the right.The concentration (C′) of an agonist required to produce a given
effect in the presence of a fixed concentration ([I]) of com-petitive
antagonist is greater than the agonist concentration (C) required to produce
the same effect in the absence of the antago-nist. The ratio of these two
agonist concentrations (dose ratio) is related to the dissociation constant (Ki)
of the antagonist by the Schild equation:
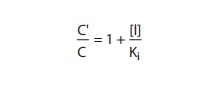
Pharmacologists
often use this relation to determine the Ki of a competitive antagonist.
Even without knowledge of the relation between agonist occupancy of the
receptor and response, the Ki can be determined simply and
accurately. As shown in Figure 2–3, concentration-response curves are obtained
in the presence and in the absence of a fixed concentration of competitive
antagonist; comparison of the agonist concentrations required to produce
identical degrees of pharmacologic effect in the two situations reveals the
antagonist’s Ki . If C′ is twice C, for example, then [I]= Ki.
For
the clinician, this mathematical relation has two important therapeutic
implications:
1.
The degree of inhibition produced by a competitive antagonist depends on the
concentration of antagonist. The competitive β-adrenoceptor antagonist propranolol provides
a usefulexample. Patients receiving a fixed dose of this drug exhibit a wide
range of plasma concentrations, owing to differences among individuals in
clearance of propranolol. As a result, inhibitory effects on physiologic
responses to norepinephrine and epinephrine (endogenous adrenergic receptor
agonists) may vary widely, and the dose of propranolol must be adjusted
accordingly.
2.
Clinical response to a competitive antagonist also depends on the concentration
of agonist that is competing for binding to receptors. Again, propranolol
provides a useful example: When this drug is administered at moderate doses
sufficient to block the effect of basal levels of the neurotransmitter
norepinephrine, resting heart rate is decreased. However, the increase in the
release of norepinephrine and epinephrine that occurs with exercise, postural
changes, or emotional stress may suffice to overcome this competitive
antagonism. Accordingly, the same dose of propranolol may have little effect
under these condi-tions, thereby altering therapeutic response.
Some
receptor antagonists bind to the receptor in an irrevers-ible or nearly irreversible fashion, either by forming a
covalentbond with the receptor or by binding so tightly that, for practical
purposes, the receptor is unavailable for binding of agonist. After occupancy
of some proportion of receptors by such an antagonist, the number of remaining
unoccupied receptors may be too low for the agonist (even at high concentrations)
to elicit a response com-parable to the previous maximal response (Figure
2–3B). If spare receptors are present, however, a lower dose of an irreversible
antagonist may leave enough receptors unoccupied to allowachievement of maximum
response to agonist, although a higher agonist concentration will be required
(Figure 2–2B and C; see Receptor-Effector Coupling & Spare Receptors).
Therapeutically,
irreversible antagonists present distinct advan-tages and disadvantages. Once
the irreversible antagonist has occupied the receptor, it need not be present
in unbound form to inhibit agonist responses. Consequently, the duration of
action of such an irreversible antagonist is relatively independent of its own
rate of elimination and more dependent on the rate of turnover of receptor
molecules.
Phenoxybenzamine,
an irreversible α-adrenoceptor
antagonist, is used to control the hypertension caused by catecholamines
released from pheochromocytoma, a tumor of the adrenal medulla. If
administration of phenoxybenzamine lowers blood pressure, blockade will be
maintained even when the tumor epi-sodically releases very large amounts of
catecholamine. In this case, the ability to prevent responses to varying and
high concen-trations of agonist is a therapeutic advantage. If overdose occurs,
however, a real problem may arise. If the α-adrenoceptor blockade cannot be overcome,
excess effects of the drug must be antago-nized “physiologically,” ie, by using
a pressor agent that does not act via α receptors.
Antagonists
can function noncompetitively in a different way; that is, by binding to a site
on the receptor protein separate from the agonist binding site, and thereby
modifying receptor activity without blocking agonist binding (see Figure 1–3C
and D). Although these drugs act noncompetitively, their actions are reversible
if they do not bind covalently. Such drugs are often called allosteric modulators. For example,
benzodiazepines bind noncompetitively to ion channels activated by the
neurotransmit-ter γ-aminobutyric acid (GABA),
enhancing the net activating effect of GABA on channel conductance.
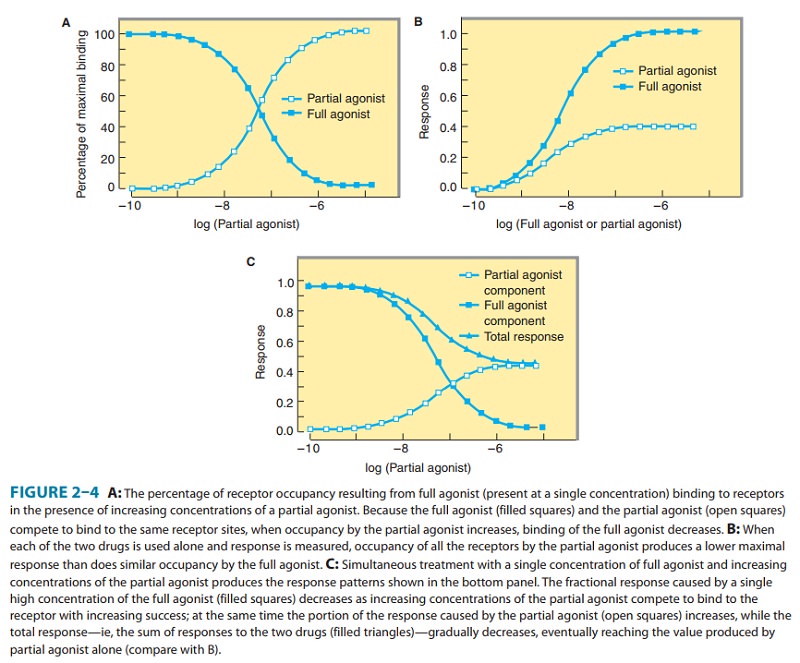
Partial Agonists
Based
on the maximal pharmacologic response that occurs when all receptors are
occupied, agonists can be divided into two classes: partial agonists produce a lower response, at full receptor
occu-pancy, than do full agonists.
Partial agonists produce concentra-tion-effect curves that resemble those
observed with full agonists in the presence of an antagonist that irreversibly
blocks some of the receptor sites (compare Figures 2–2 [curve D] and 2–4B). It
is important to emphasize that the failure of partial agonists to pro-duce a
maximal response is not due to decreased affinity for binding to receptors.
Indeed, a partial agonist’s inability to cause a maximal pharmacologic
response, even when present at high concentrations that saturate binding to all
receptors, is indicated by the fact that partial agonists competitively inhibit
the responses produced by full agonists (Figure 2–4C). Many drugs used
clinically as antagonists are actually weak partial agonists. Partial agonism
can be useful in some clinical circumstances. For example, buprenorphine, a
partial agonist of μ-opioid receptors, is a generally safer analgesic drug than
mor-phine because it produces less respiratory depression in overdose.
Buprenorphine is effectively antianalgesic when administered to
morphine-dependent individuals, however, and may precipitate a drug withdrawal
syndrome due to competitive inhibition of mor-phine’s agonist action.
Other Mechanisms of Drug Antagonism
Not
all the mechanisms of antagonism involve interactions of drugs or endogenous
ligands at a single type of receptor, and some types of antagonism do not
involve a receptor at all. For example, protamine, a protein that is positively
charged at physiologic pH, can be used clinically to counteract the effects of
heparin, an anti-coagulant that is negatively charged. In this case, one drug
acts as a chemical antagonist of the
other simply by ionic binding that makes the other drug unavailable for
interactions with proteins involved in blood clotting.
Another
type of antagonism is physiologic
antagonism between endogenous regulatory pathways mediated by different
receptors. For example, several catabolic actions of the glucocorticoidhormones
lead to increased blood sugar, an effect that is physio-logically opposed by
insulin. Although glucocorticoids and insulin act on quite distinct
receptor-effector systems, the clinician must sometimes administer insulin to
oppose the hyperglycemic effects of a glucocorticoid hormone, whether the
latter is elevated by endogenous synthesis (eg, a tumor of the adrenal cortex)
or as a result of glucocorticoid therapy.
In
general, use of a drug as a physiologic antagonist produces effects that are
less specific and less easy to control than are the effects of a
receptor-specific antagonist. Thus, for example, to treat bradycardia caused by
increased release of acetylcholine from vagus nerve endings, the physician
could use isoproterenol, a β-adrenoceptor agonist that increases heart
rate by mimickingsympathetic stimulation of the heart. However, use of this
physi-ologic antagonist would be less rational—and potentially more
dangerous—than would use of a receptor-specific antagonist such as atropine (a
competitive antagonist at the receptors at which acetylcholine slows heart
rate).
Related Topics