Chapter: Medical Surgical Nursing: Management of Patients With Chest and Lower Respiratory Tract Disorders
Pulmonary Hypertension
Pulmonary Hypertension
Pulmonary
hypertension is a condition that is not clinically evi-dent until late in its
progression. Pulmonary hypertension exists when the systolic pulmonary artery
pressure exceeds 30 mm Hg or the mean pulmonary artery pressure exceeds 25 mm
Hg. These pressures cannot be measured indirectly as can systemic blood
pressure; instead, they must be measured during right-sided heart
catheterization. In the absence of these measurements, clinical recognition
becomes the only indicator for the presence of pul-monary hypertension.
There
are two forms of pulmonary hypertension: primary (or idiopathic) and secondary.
Primary pulmonary hypertension is an uncommon disease in which the diagnosis is
made by exclud-ing all other possible causes. The exact cause is unknown, but
there are several possible causes (Chart 23-7). The clinical pre-sentation of
primary pulmonary hypertension exists with no evi-dence of pulmonary and
cardiac disease or pulmonary embolism. It occurs most often in women 20 to 40
years of age and is usu-ally fatal within 5 years of diagnosis.
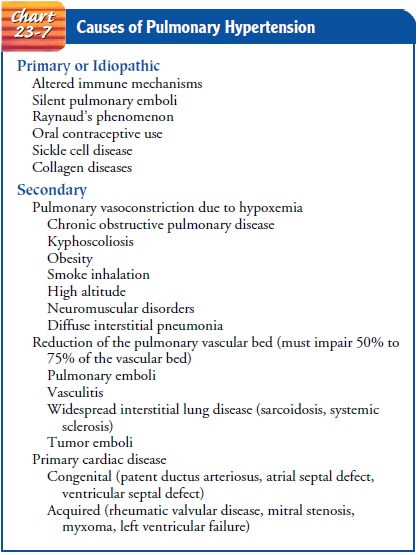
Secondary
pulmonary hypertension is more common and re-sults from existing cardiac or
pulmonary disease. The prognosis depends on the severity of the underlying
disorder and the changes in the pulmonary vascular bed. A common cause of
secondary pul-monary hypertension is pulmonary artery constriction due to
hy-poxemia from COPD.
Pathophysiology
The
underlying process of pulmonary hypertension varies, and multiple factors are
often responsible. Normally, the pulmonary vascular bed can handle the blood
volume delivered by the right ventricle. It has a low resistance to blood flow
and compensates for increased blood volume by dilation of the vessels in the
pulmonary circulation. However, if the pulmonary vascular bed is destroyed or
obstructed, as in pulmonary hypertension, the ability to handle whatever flow
or volume of blood it receives is impaired, and the increased blood flow then
increases the pulmonary artery pressure. As the pulmonary arterial pressure
increases, the pulmonary vas-cular resistance also increases. Both pulmonary
artery constriction (as in hypoxemia or hypercapnia) and a reduction of the
pul-monary vascular bed (which occurs with pulmonary emboli) re-sult in an
increase in pulmonary vascular resistance and pressure. This increased workload
affects right ventricular function. The myocardium ultimately cannot meet the
increasing demands im-posed on it, leading to right ventricular hypertrophy
(enlargement and dilation) and failure.
Clinical Manifestations
Dyspnea
is the main symptom of pulmonary hypertension, oc-curring at first with
exertion and eventually at rest. Substernal chest pain also is common,
affecting 25% to 50% of patients. Other signs and symptoms include weakness,
fatigue, syncope, occasional hemoptysis, and signs of right-sided heart failure
(pe-ripheral edema, ascites, distended neck veins, liver engorgement, crackles,
heart murmur).
Assessment and Diagnostic Findings
A
complete diagnostic evaluation includes a history, physical ex-amination, chest
x-ray, pulmonary function studies, electrocar-diogram (ECG), echocardiogram,
ventilation–perfusion scan, and cardiac catheterization. In some cases, a lung
biopsy, performed by thoracotomy or thoracoscopy, may be needed to make a
defi-nite diagnosis. Cardiac catheterization of the right side of the heart
reveals elevated pulmonary arterial pressure. An echocardiogram can assess the
progression of the disease and rule out other condi-tions with similar signs
and symptoms. The ECG reveals right ventricular hypertrophy, right axis
deviation, and tall peaked P waves in inferior leads, tall anterior R waves,
and ST-segment depression and/or T-wave inversion anteriorly. The PaO2
also is decreased (hypoxemia). A ventilation–perfusion scan or pul-monary
angiography detects defects in pulmonary vasculature, such as pulmonary emboli.
Pulmonary function studies may be normal or show a slight decrease in vital
capacity (VC) and lung compliance, with a mild decrease in the diffusing
capacity.
Medical Management
The
goal of treatment is to manage the underlying cardiac or pul-monary condition.
Most patients with primary pulmonary hyper-tension do not have hypoxemia at
rest but require supplemental oxygen with exercise. However, patients with
severe right ventric-ular failure, decreased cardiac output, and progressive
disease may have resting hypoxemia and require continuous oxygen
supple-mentation. Appropriate oxygen therapy reverses the vasoconstriction and
reduces the pulmonary hypertension in a rel-atively short time.
In
the presence of cor pulmonale, which is discussed in the section that follows,
treatment should include fluid restriction, diuretics to decrease fluid
accumulation, cardiac glycosides (eg, digitalis) in an attempt to improve
cardiac function, calcium channel blockers for vasodilation, and rest. In
primary pulmonary hypertension, vasodilators have been administered with
variable success (eg, calcium channel blockers, intravenous prostacyclin).
Prostacyclin (PGX [Flolan]) is one of the prostaglandins pro-duced by the
pulmonary endothelium. Intravenous prostacyclin (epoprostenol) helps to
decrease pulmonary hypertension by re-ducing pulmonary vascular resistance and
pressures and increas-ing cardiac output. Anticoagulants such as warfarin
(Coumadin) have been given to patients because of chronic pulmonary em-boli.
Heart– lung transplantation has been successful in select pa-tients with
primary hypertension who have not been responsive to other therapies.
Nursing Management
The
major nursing goal is to identify patients at high risk for pul-monary
hypertension, such as those with COPD, pulmonary emboli, congenital heart
disease, and mitral valve disease. The nurse also must be alert for signs and
symptoms, administer oxy-gen therapy appropriately, and instruct patients and
their fami-lies about the use of home oxygen supplementation.
Related Topics