Chapter: Basic & Clinical Pharmacology : Pharmacologic Management of Parkinsonism & Other Movement Disorders
Parkinsonism
PARKINSONISM
Parkinsonism is
characterized by a combination of rigidity, bradykinesia, tremor, and postural
instability that can occur for a variety of reasons but is usually idiopathic (Parkinson’s
disease or paralysis agitans). Cognitive decline occurs in many patients as the
disease advances. Other nonmotor symptoms—which are receiving increasing
attention—are affective disorders (anxiety or depression), personality changes,
abnormalities of autonomic function (sphincter or sexual functions; choking;
sweating abnor-malities; and disturbances of blood pressure regulation), sleep
disorders, and sensory complaints or pain. The disease is generally
progressive, leading to increasing disability unless effective treatment is
provided.
Pathogenesis
The pathogenesis of
parkinsonism seems to relate to a combina-tion of impaired degradation of
proteins, intracellular protein accumulation and aggregation, oxidative stress,
mitochondrial damage, inflammatory cascades, and apoptosis. Studies in twins
suggest that genetic factors are important, especially when the disease occurs
in patients under age 50. Recognized genetic abnor-malities account for 10–15%
of cases. Mutations of the α-synuclein gene at 4q21 or duplication and
triplication of the normal synu-clein gene are associated with Parkinson’s
disease, which is now widely recognized as a synucleinopathy. Mutations of the leucine-rich repeat kinase 2 (LRRK2) gene at 12cen, and the UCHL1 gene may also cause autosomal
dominant parkinsonism. Mutations in the parkin
gene (6q25.2–q27) cause early-onset, autosomal reces-sive, familial
parkinsonism, or sporadic juvenile-onset parkin-sonism. Several other genes or
chromosomal regions have been associated with familial forms of the disease.
Environmental or endogenous toxins may also be important in the etiology of the
disease. Epidemiologic studies reveal that cigarette smoking, cof-fee,
anti-inflammatory drug use, and high serum uric acid levels are protective,
whereas the incidence of the disease is increased in those working in teaching,
health care, or farming, and in those with lead or manganese exposure or with
vitamin D deficiency.The finding of Lewy bodies (intracellular inclusion bodies
containing α-synuclein)
in fetal dopaminergic cells transplanted into the brain of parkinsonian
patients some years previously has provided some support for suggestions that
Parkinson’s disease may represent a prion disease.Staining for α-synuclein has
revealed that pathology is more widespread than previously recognized,
developing initially in the olfactory nucleus and lower brainstem (stage 1 of
Braak), then the higher brainstem (stage 2), the substantia nigra (stage 3),
the mesocortex and thalamus (stage 4), and finally the entire neo-cortex (stage
5). The motor features of Parkinson’s disease develop at stage 3 on the Braak
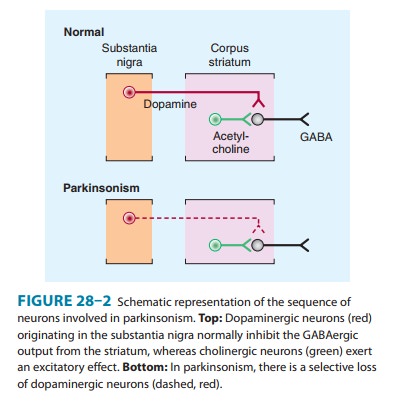
scale.The normally high concentration of dopamine in the basal ganglia of the
brain is reduced in parkinsonism, and pharmaco-logic attempts to restore
dopaminergic activity with levodopa and dopamine agonists alleviate many of the
motor features of the disorder. An alternative but complementary approach has
been to restore the normal balance of cholinergic and dopaminergic influ-ences
on the basal ganglia with antimuscarinic drugs. The pathophysiologic basis for
these therapies is that in idiopathic parkinsonism, dopaminergic neurons in the
substantia nigra that normally inhibit the output of GABAergic cells in the
corpus striatum are lost (Figure 28–2). Drugs that induce parkinsonian
syndromes either are dopamine receptor antagonists (eg, antipsy-chotic agents;)
or lead to the destruction of the dopaminergic nigrostriatal neurons (eg,
1-methyl-4-phenyl-1-,2,3,6-tetrahydropyridine [MPTP]; ). Various other
neurotransmitters, such as norepinephrine, are also depleted in the brain in
parkinsonism, but these deficiencies are of uncertain clinical relevance.
Related Topics