Chapter: Basic & Clinical Pharmacology : Pharmacologic Management of Parkinsonism & Other Movement Disorders
Other Movement Disorders
OTHER MOVEMENT DISORDERS
Tremor
Tremor consists of
rhythmic oscillatory movements. Physiologic postural tremor, which is a normal
phenomenon, is enhanced in amplitude by anxiety, fatigue, thyrotoxicosis, and
intravenous epinephrine or isoproterenol. Propranolol
reduces its amplitude and, if administered intra-arterially, prevents the
response to iso-proterenol in the perfused limb, presumably through some
peripheral action. Certain drugs—especially the bronchodilators, valproate,
tricyclic antidepressants, and lithium—may produce a dose-dependent
exaggeration of the normal physiologic tremor that is reversed by discontinuing
the drug. Although the tremor produced by sympathomimetics such as terbutaline
(a bronchodi-lator) is blocked by propranolol, which antagonizes both β1 and β2receptors, it is not
blocked by metoprolol, a β1-selective antag-onist; this suggests that
such tremor is mediated mainly by the β2 receptors.
Essential tremor is a
postural tremor, sometimes familial with autosomal dominant inheritance, which
is clinically similar to physiologic tremor. At least three gene loci (ETM1 on 3q13, ETM2 on 2p24.1, and a locus on 6p23) have been described.
Dysfunction of β1 receptors has been implicated in some instances, since the
tremor may respond dramatically to standard doses of metoprolol as well as to
propranolol. The most useful approach is with propranolol, but whether the
response depends on a central or peripheral action is unclear.
The pharmacokinetics,
pharmaco-logic effects, and adverse reactions of propranolol are discussed.
Daily doses of propranolol on the order of 120 mg (range, 60–240 mg) are
usually required, prescribed as 40–120 mg orally twice daily, and reported
adverse effects have been few. Propranolol should be used with caution in
patients with heart failure, heart block, asthma, and hypoglycemia. Patients
can be instructed to take their own pulse and call the physician if
signifi-cant bradycardia develops. Metoprolol is sometimes useful in treating
tremor when patients have concomitant pulmonary dis-ease that contraindicates
use of propranolol. Primidone (an
anti-epileptic drug;), in gradually increasing doses up to 250 mg three times
daily, is also effective in providing symp-tomatic control in some cases.
Patients with tremor are very sensi-tive to primidone and often cannot tolerate
the doses used to treat seizures; they should be started on 50 mg once daily
and the daily dose increased by 50 mg every 2 weeks depending on response.
Topiramate, another antiepileptic drug, may also be helpful ina dose of
400 mg daily, built up gradually. Alprazolam
(in doses up to 3 mg daily) or gabapentin
(100–2400 mg/d) is helpful in some patients. Others are helped by intramuscular
injections of botulinum toxin. Thalamic stimulation by an implanted electrode
and stimulator is often worthwhile in advanced cases refractory to
pharmacotherapy. Diazepam, chlordiazepoxide, mephenesin, and antiparkinsonism
agents have been advocated in the past but are generally worthless. Anecdotal
reports of benefit from mirtazapine were not confirmed in a double-blind study,
which found no effect on the tremor in most patients. Small quantities of
alcohol may suppress essential tremor for a short time but should not be
recommended as a treatment strategy because of possible behav-ioral and other
complications of alcohol.
Intention tremor is present during movement but not at rest;sometimes it occurs
as a toxic manifestation of alcohol or drugs such as phenytoin. Withdrawal or
reduction in dosage provides dramatic relief. There is no satisfactory
pharmacologic treatment for intention tremor due to other neurologic disorders.
Rest tremor is usually due to parkinsonism.
Huntington’s Disease
Huntington’s disease
is an autosomal dominant inherited disorder caused by an abnormality (expansion
of a CAG trinucleotide repeat that codes for a polyglutamine tract) of the huntingtin gene on chromosome 4. An
autosomal recessive form may also occur. Huntington disease–like (HDL)
disorders are not associated with an abnormal CAG trinucleotide repeat number
of the huntingtin gene. Autosomal
dominant (HDL1, 20pter-p12; HDL2, 16q24.3) and recessive forms (HDL3, 4p15.3) occur.
Huntington’s disease
is characterized by progressive chorea and dementia that usually begin in
adulthood. The development of chorea seems to be related to an imbalance of
dopamine, acetyl-choline, GABA, and perhaps other neurotransmitters in the
basal ganglia (Figure 28–6). Pharmacologic studies indicate that chorea results
from functional overactivity in dopaminergic nigrostriatal pathways, perhaps
because of increased responsiveness of post-synaptic dopamine receptors or
deficiency of a neurotransmitter
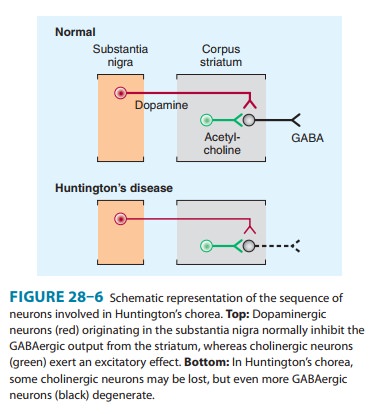
Drugs that impair dopamin-ergic neurotransmission, either by
depleting central monoamines (eg, reserpine, tetrabenazine) or by blocking
dopamine receptors (eg, phenothiazines, butyrophenones), often alleviate
chorea, whereas dopamine-like drugs such as levodopa tend to exacerbate it.
Both GABA and the
enzyme (glutamic acid decarboxylase) concerned with its synthesis are markedly
reduced in the basal ganglia of patients with Huntington’s disease, and GABA
recep-tors are usually implicated in inhibitory pathways. There is also a
significant decline in concentration of choline acetyltransferase, the enzyme
responsible for synthesizing acetylcholine, in the basal ganglia of these
patients. These findings may be of pathophysio-logic significance and have led
to attempts to alleviate chorea by enhancing central GABA or acetylcholine
activity, but with disap-pointing results. As a consequence, the most commonly
used drugs for controlling dyskinesia in patients with Huntington’s disease are
still those that interfere with dopamine activity. With all the latter drugs,
however, reduction of abnormal movements may be associated with iatrogenic
parkinsonism.
Reserpine depletes cerebral dopamine by preventing intraneu-ronal storage
; it is introduced in low doses (eg, 0.25 mg daily), and the daily dose is then
built up gradually (eg, by 0.25 mg every week) until benefit occurs or adverse
effects become troublesome. A daily dose of 2–5 mg is often effective in
suppressing abnormal movements, but adverse effects may include hypotension,
depression, sedation, diarrhea, and nasal congestion. Tetrabenazine (12.5–50 mg orally three times daily)
resemblesreserpine in depleting cerebral dopamine and has less troublesome
adverse effects. Treatment with postsynaptic dopamine receptor blockers such as
phenothiazines and butyrophenones may also be helpful. Haloperidol
is started in a small dose, eg, 1 mg twice daily, and increased every 4 days depending on the
response. If haloperi-dol is not helpful, treatment with increasing doses of perphenazine up to a total of about 20
mg daily sometimes helps. Several recent reports suggest that olanzapine may also be useful; the dose
varies with the patient, but 10 mg daily is often sufficient, although doses as
high as 30 mg daily are sometimes required. The pharmacoki-netics and clinical
properties of these drugs are considered in greater detail elsewhere in this
book. Selective serotonin reuptake inhibitors may reduce depression,
aggression, and agitation.
Other Forms of Chorea
Benign hereditary
chorea is inherited (usually autosomal domi-nant; possibly also autosomal
recessive) or arises spontaneously. Chorea develops in early childhood and does
not progress during adult life; dementia does not occur. In patients with TITF-1 gene mutations, thyroid and
pulmonary abnormalities may also be pres-ent (brain-thyroid-lung syndrome).
Familial chorea may also occur as part of the chorea-acanthocytosis syndrome,
together with oro-lingual tics, vocalizations, cognitive changes, seizures,
peripheral neuropathy, and muscle atrophy; serum β-lipoproteins are normal. Mutations of the
gene encoding chorein at 9q21 may be causal. Treatment of these hereditary
disorders is symptomatic.
Treatment is directed
at the underlying cause when chorea occurs as a complication of general medical
disorders such as thy-rotoxicosis, polycythemia vera rubra, systemic lupus
erythemato-sus, hypocalcemia, and hepatic cirrhosis. Drug-induced chorea is
managed by withdrawal of the offending substance, which may be levodopa, an
antimuscarinic drug, amphetamine, lithium, pheny-toin, or an oral
contraceptive. Neuroleptic drugs may also produce an acute or tardive dyskinesia
(discussed below). Sydenham’s cho-rea is temporary and usually so mild that
pharmacologic manage-ment of the dyskinesia is unnecessary, but
dopamine-blocking drugs are effective in suppressing it.
Ballismus
The biochemical basis
of ballismus is unknown, but the pharma-cologic approach to management is the
same as for chorea. Treatment with haloperidol, perphenazine, or other
dopamine-blocking drugs may be helpful.
Athetosis & Dystonia
The pharmacologic
basis of these disorders is unknown, and there is no satisfactory medical
treatment for them. A subset of patients respond well to levodopa medication
(dopa-responsive dystonia), which is therefore worthy of trial. Occasional
patients with dysto-nia may respond to diazepam, amantadine, antimuscarinic drugs
(in high dosage), carbamazepine, baclofen, haloperidol, or phe-nothiazines. A
trial of these pharmacologic approaches is worth-while, though often not
successful. Patients with focal dystonias such as blepharospasm or torticollis
often benefit from injection of botulinum toxin into the overactive muscles.
Deep brain stimu-lation may be helpful in medically intractable cases.
Tics
The pathophysiologic
basis of tics is unknown. Chronic multiple tics (Gilles de la Tourette’s syndrome) may require symptomatic treatment
if the disorder is severe or is having a significant impact on the patient’s
life. Education of patients, family, and teachers is important.
A common pharmacologic
approach is with haloperidol.
Patients are better able to tolerate this drug if treatment is started with a
small dosage (eg, 0.25 or 0.5 mg daily) and then increased gradually (eg, by
0.25 mg every 4 or 5 days) over the following weeks depending on response and
tolerance. Most patients ulti-mately require a total daily dose of 3–8 mg.
Adverse effects include extrapyramidal movement disorders, sedation, dryness of
the mouth, blurred vision, and gastrointestinal disturbances. Pimozide, another dopamine receptor
antagonist, may be helpfulin patients as a first-line treatment or in those who
are either unre-sponsive to or intolerant of haloperidol. Treatment is started
at 1 mg/d, and the dosage is increased by 1 mg every 5 days; most patients
require 7–16 mg/d. It has similar side effects to haloperi-dol but may cause
irregularities of cardiac rhythm.
Although not approved
by the FDA for the treatment of tics or Tourette’s syndrome, certain α-adrenergic agonists
may be pre-ferred as an initial treatment because they are less likely to cause
extrapyramidal side effects than neuroleptics agents. Clonidine reduces motor or vocal tics in about 50% of children so
treated. It may act by reducing activity in noradrenergic neurons in the locus
caeruleus. It is introduced at a dose of 2–3 mcg/kg/d, increasing after 2 weeks
to 4 mcg/kg/d and then, if required, to 5 mcg/kg/d. It may cause an initial
transient fall in blood pressure. The most common adverse effect is sedation;
other adverse effects include reduced or excessive salivation and diarrhea. Guanfacine, another α-adrenergic agonist,
has also been used.
Phenothiazines such as
fluphenazine sometimes help the tics, as do dopamine agonists. Atypical
antipsychotics, such as risperi-done and aripiprazole, have a more favorable
side-effect profile and may be especially worthwhile in patients with
significant behavioral problems. Clonazepam and carbamazepine have also been
used. The pharmacologic properties of these drugs are dis-cussed elsewhere in
this book.
Injection of botulinum
toxin A at the site of problematic tics is sometimes helpful. Treatment of any
associated attention deficit disorder (eg, with clonidine patch, guanfacine,
pemoline, meth-ylphenidate, or dextroamphetamine) or obsessive-compulsive
dis-order (selective serotonin reuptake inhibitors or clomipramine) may be
required. Deep brain stimulation is sometimes worthwhile in otherwise
intractable cases but is best regarded as an investiga-tional approach at this
time.
Drug-Induced Dyskinesias
Levodopa or dopamine
agonists produce diverse dyskinesias as a dose-related phenomenon in patients
with Parkinson’s disease; dose reduction reverses them. Chorea may also develop
in patients receiving phenytoin, carbamazepine, amphetamines, lithium, and oral
contraceptives, and it resolves with discontinuance of the offending
medication. Dystonia has resulted from administrationof dopaminergic agents,
lithium, serotonin reuptake inhibitors, carbamazepine, and metoclopramide; and
postural tremor from theophylline, caffeine, lithium, valproic acid, thyroid
hormone, tricyclic antidepressants, and isoproterenol.
The pharmacologic
basis of the acute dyskinesia or dystonia sometimes precipitated by the first
few doses of a phenothiazine is not clear. In most instances, parenteral
administration of an anti-muscarinic drug such as benztropine (2 mg
intravenously), diphenhydramine (50 mg intravenously), or biperiden (2–5 mg
intravenously or intramuscularly) is helpful, whereas in other instances
diazepam (10 mg intravenously) alleviates the abnormal movements.
Tardive dyskinesia,a disorder
characterized by a variety ofabnormal movements, is a common complication of
long-term neuroleptic or metoclopramide drug treatment . Its precise
pharmacologic basis is unclear. A reduction in dose of the offending
medication, a dopamine receptor blocker, commonly worsens the dyskinesia,
whereas an increase in dose may suppress it. The drugs most likely to provide
immediate symptomatic benefit are those interfering with dopaminergic function,
either by deple-tion (eg, reserpine, tetrabenazine) or receptor blockade (eg,
phe-nothiazines, butyrophenones). Paradoxically, the receptor-blocking drugs
are the very ones that also cause the dyskinesia.
Tardive dystonia is usually segmental or focal; generalizeddystonia is less
common and occurs in younger patients. Treatment is the same as for tardive
dyskinesia, but anticholin-ergic drugs may also be helpful; focal dystonias may
also respond to local injection of botulinum A toxin. Tardive akathisia is treated similarly to drug-induced parkinsonism.
Rabbit syn-drome, another
neuroleptic-induced disorder, is manifested byrhythmic vertical movements about
the mouth; it may respond to anticholinergic drugs.
Because the tardive
syndromes that develop in adults are often irreversible and have no satisfactory
treatment, care must be taken to reduce the likelihood of their occurrence.
Antipsychotic medication should be prescribed only when neces-sary and should
be withheld periodically to assess the need for continued treatment and to
unmask incipient dyskinesia. Thioridazine, a phenothiazine with a piperidine
side chain, is an effective antipsychotic agent that seems less likely than
most to cause extrapyramidal reactions, perhaps because it has little effect on
dopamine receptors in the striatal system. Finally, anti-muscarinic drugs
should not be prescribed routinely in patients receiving neuroleptics, because
the combination may increase the likelihood of dyskinesia.
Neuroleptic malignant syndrome, a rare complication of treat-ment with
neuroleptics, is characterized by rigidity, fever, changes in mental status,
and autonomic dysfunction (see Table 16–4). Symptoms typically develop over 1–3
days (rather than minutes to hours as in malignant hyperthermia) and may occur
at any time during treatment. Treatment includes withdrawal of antipsychotic
drugs, lithium, and anticholinergics; reduction of body tempera-ture; and
rehydration. Dantrolene, dopamine agonists, levodopa, or amantadine may be
helpful, but there is a high mortality rate (up to 20%) with neuroleptic
malignant syndrome.
Restless Legs Syndrome
Restless legs syndrome
is characterized by an unpleasant creeping discomfort that seems to arise deep
within the legs and occasion-ally the arms. Symptoms occur particularly when
patients are relaxed, especially when they are lying down or sitting, and they
lead to an urge to move about. Such symptoms may delay the onset of sleep. A
sleep disorder associated with periodic move-ments during sleep may also occur.
The cause is unknown, but the disorder is especially common among pregnant
women and also among uremic or diabetic patients with neuropathy. In most
patients, no obvious predisposing cause is found, but several genetic loci have
been associated with it (12q12-q21, 14q13-q31, 9p24-p22, 2q33, and 20p13).
Symptoms may resolve
with correction of coexisting iron-deficiency anemia and often respond to
dopamine agonists, levodopa, diazepam, clonazepam, gabapentin, or opiates.
Dopaminergic therapy is the preferred treatment for restless legs syndrome and should
be initiated with long-acting dopamine agonists (eg, pramipexole 0.125–0.75 mg or ropinirole
0.25–4.0 mg once daily) to avoid the augmentation that may be associated with
levodopa-carbidopa (100/25 or 200/50 taken about 1 hour before bedtime). Augmentation
refers to the earlier onset or enhancement of symptoms; earlier onset of
symptoms at rest; and a briefer response to medication. When augmentation
occurs with levodopa, the daily dose should be reduced or a dopamine agonist
substituted. If it occurs in patients receiving an agonist, the daily dose
should be lowered or divided, or opioids substituted. When opiates are
required, those with long half-lives or low addictive potential should be used.
Oxycodone is often effective; the dose is individualized. Gabapentin is an
alternative to opioids and is taken once or twice daily (in the evening and
before sleep); the starting dose is 300 mg daily, building up depending on
response and tolerance (to approximately 1800 mg daily). A recent study
suggests that pregabalin, a related drug, is also effective at a daily total
dosage of 150–300 mg, taken in divided doses.
Wilson’s Disease
A recessively
inherited (13q14.3–q21.1) disorder of copper metabolism, Wilson’s disease is
characterized biochemically by reduced serum copper and ceruloplasmin
concentrations, patho-logically by markedly increased concentration of copper
in the brain and viscera, and clinically by signs of hepatic and neurologic
dysfunction. Neurologic signs include tremor, choreiform move-ments, rigidity,
hypokinesia, and dysarthria and dysphagia. Siblings of affected patients should
be screened for asymptomatic Wilson’s disease.
Treatment involves the
removal of excess copper, followed by maintenance of copper balance. Dietary
copper should also be kept below 2 mg daily. Penicillamine (dimethylcysteine) has been used for many years as
the primary agent to remove copper. It is a chelating agent that forms a ring
complex with copper. It is readily absorbed from the gastrointestinal tract and
rapidly excreted in the urine. A common starting dose in adults is 500 mg three
or four times daily. After remission occurs, it may be possible to lower the
maintenance dose, generally to not less than 1 g daily, which must thereafter
be continued indefinitely. Adverse effects include nausea and vomiting,
nephrotic syndrome, a lupus-like syndrome, pem-phigus, myasthenia, arthropathy,
optic neuropathy, and various blood dyscrasias. In about 10% of instances,
neurologic worsening occurs with penicillamine. Treatment should be monitored
by frequent urinalysis and complete blood counts.
Trientine hydrochloride,another chelating
agent, is preferred bymany over penicillamine because of the lesser likelihood
of drug reac-tions or neurologic worsening. It may be used in a daily dose of
1–1.5 g. Trientine appears to have few adverse effects other than mild anemia due
to iron deficiency in a few patients. Tetrathiomolybdate
may be better than trientine for preserving neurologic function in patients with
neurologic involvement and is taken both with and between meals. It is not yet
commercially available.
Zinc acetate administered
orally increases the fecal excretion of copper and can be used in combination
with these other agents. The dose is 50 mg three times a day. Zinc sulfate (200
mg/d orally) has also been used to decrease copper absorption. Zinc blocks
copper absorption from the gastrointestinal tract by induc-tion of intestinal
cell metallothionein. Its main advantage is its low toxicity compared with that
of other anticopper agents, although it may cause gastric irritation when
introduced.
Liver transplantation
is sometimes necessary. The role of hepatocyte transplantation and gene therapy
is currently under investigation.
Related Topics