Chapter: Modern Medical Toxicology: Miscellaneous Drugs and Poisons: Gastrointestinal and Endocrinal Drugs
Immunomodulators
IMMUNOMODULATOR
Immunosuppressive Agents
Cyclosporine (Cyclosporin A)
Cyclosporine
belongs to a family of cyclic polypeptides derived from the fungus Tolypocladium inflatum Gams. It is
lipophilic and hydrophobic, and therefore must be solubilised for clinical use.
Uses
·
To prevent transplant rejection in the transplantation of
kidney, heart, and liver. Cyclosporine is usually combined with
corti-costeroids. It is also being increasingly used in transplantation of
other organs such as lung, pancreas, and bone marrow.
·
Cyclosporine is also beneficial in the treatment of
psoriasis, rheumatoid arthritis, Crohn’s disease, nephrotic syndrome,
endogenous uveitis, atopic dermatitis, and acute ocular Behcet’s syndrome.
·
There are also indications that cyclosporine may be useful
in the treatment of primary biliary cirrhosis, pyoderma gangrenosum,
polymyositis, aplastic anaemia, myasthenia gravis and severe asthma.
Toxicokinetics
Cyclosporine
can be administered orally, intravenously, or by injection. When given orally,
it is metabolised on first pass through the liver, its absolute bioavailability
being about 35%. Peak plasma concentration occurs at about 2.5 hours. About 50%
of the drug in whole blood is bound to erythrocytes. The apparent volume of
distribution in adults is 4.7 L/kg. Elimination occurs predominantly by
metabolism in the liver by cytochrome P450 III A oxidase, and only about 0.1%
of a dose is excreted unchanged.
For
therapeutic purposes, cyclosporine levels in plasma should not exceed 150 ng/ml
(600 ng/ml in whole blood).
Mode of Action
·
Inhibition of T-lymphocyte
proliferation.
·
Inhibition (reversible) of
activation of primary helper T cell.
·
Decreases production and secretion
of interleukin-2.
·
Inhibition of production of
interferon gamma by lymphocytes.
Adverse Effects
·
CNS: Tremor, palmar and plantar
paraesthesia, headache, flushing, depression, visual disorders, convulsions.
·
GIT: Anorexia, nausea, vomiting,
acute pancreatitis (rare).
·
Hepatic: Cholestasis with
hyperbilirubinaemia.
·
Renal: Nephropathy can occur in up
to 75% of patients, and is the most consistent and serious of the adverse
effects.
·
CVS: Hypertension.
·
Other effects: Hypertrichosis,
gingival hyperplasia, hyper- glycaemia, hyperkalaemia, gynaecomastia,
myopathies, increased susceptibility to infections.
Drug Interactions
Nephrotoxicity
is greatly enhanced by concomitant admin-istration of aminoglycosides,
ciprofloxacin, cotrimoxazole, NSAIDs, colchicine and amphotericin B.
Blood
levels of cyclosporine are increased by diltiazem, doxycycline, erythromycin,
cephalosporines, ketoconazole, H2
antagonists, verapamil, and oral contraceptives, while they are decreased by
carbamazepine, isoniazid, phenobarbitone, phenytoin and rifampicin.![]()
Clinical (Toxic) Features
The
following have been reported in cyclosporine overdose (accidental and deliberate):
Headache,
nausea, vomiting, vertigo, hyperaesthesia of hands, burning sensation of feet,
abdominal pain, diarrhoea, sinus tachycardia and hypertension.
Premature
infants and neonates have developed hypoten-sion, wheezing, tachycardia,
cyanosis, metabolic acidosis, respiratory depression and renal failure.
Treatment
·
Decontamination:
Gastric lavage or emesis, activatedcharcoal, etc., may be beneficial. Multiple
dose activated charcoal produced good results in one reported case.
·
Admission to intensive care unit
followed by monitoring of vital signs and parameters.
·
Patients with stable renal function
can be treated sympto-matically and supportively. Most cases recover within 24
hours.
Tacrolimus
·
Tacrolimus is a macrolide compound
produced by Streptomycestsukubaensis.
Uses
·
Immunosuppressive agent to prevent
organ transplant rejec-tion. Tacrolimus is said to be 100 times more potent
than cyclosporine.
·
Treatment of cyclosporine-induced
haemolytic uraemic syndrome, severe psoriasis, Behcet’s disease, and Type I
diabetes mellitus.
Toxicokinetics
Tacrolimus
is poorly absorbed orally, and intravenous adminis-tration is preferred,
especially at the time of starting the course. The mean bioavailability is 25%,
and the mean apparent volume of distribution is about 19 L/kg. RBCs concentrate
tacrolimus so that whole blood values are higher than plasma values. The drug
is completely metabolised before elimination, and less than 1% of an oral or IV
dose of tacrolimus is excreted in the urine. Tacrolimus is eliminated mainly by
hepatic cytochrome P450 III A metabolism.
Mode of Action
Tacrolimus
suppresses cell-mediated and humoral responses, and is a more potent inhibitor
of lymphoproliferation than cyclosporine. It prevents the activation of T
lymphocytes in response to antigenic or mitogenic stimulation.
Adverse Effects
·
These are more pronounced with
intravenous use than with oral therapy. Common adverse effects include
insomnia, tremor,
·
![]() headache,
paraesthesia, myalgia, visual sensitivity to light, and GI distress. Serious
adverse effects include nephrotoxicity, convulsions, movement disorders,
encephalopathy, psychosis, infectious complications, hyperkalaemia and
hyperglycaemia.
headache,
paraesthesia, myalgia, visual sensitivity to light, and GI distress. Serious
adverse effects include nephrotoxicity, convulsions, movement disorders,
encephalopathy, psychosis, infectious complications, hyperkalaemia and
hyperglycaemia.
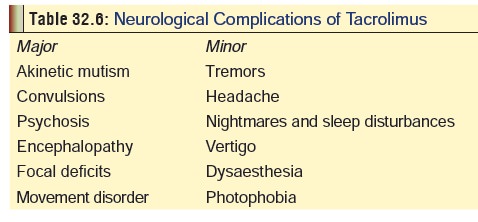
Clinical (Toxic) Features
Overdose
leads to profound immunosuppression and severe infection. Neurological
complications such as those listed in Table
32.6 are frequently seen, and generally correlate wellwith blood levels.
Treatment
Supportive
and symptomatic measures. Hyperkalaemia responds to fludrocortisone acetate.
Adrenocortical Steroids
Cytotoxic Drugs
Most
of the cytotoxic drugs have been discussed in a subsequent section ( vide infra). Only azathioprine and mycophenolate
mofetil will be discussed here.
Azathioprine (Azathioprimum)
Azathioprine
is a purine antagonist and is mainly used as an adjunct for the prevention of
kidney allografts. It is also useful in the treatment of rheumatoid arthritis.
It is invariably admin-istered orally. Azathioprine inhibits DNA synthesis, and
as a purine antagonist, exerts its effect on activated lymphocytes, which
require purines during their proliferative phase. The immunosuppressive effect
of azathioprine is believed to be due to mercaptopurine (a metabolite).
Adverse
effects include bone marrow depression, hepatic dysfunction, infection, drug
fever, nausea, vomiting, and diar-rhoea. Rash, urticaria, and vasculitis
(allergic) have also been reported. In overdose, it causes vomiting, diarrhoea,
leucopenia, hepatotoxicity.
Treatment
consists of supportive and symptomatic meas-ures. Early GI decontamination may
minimise the likelihood of bone marrow depression and hepatotoxicity.
Haemodialysis may be beneficial.
Mycophenolate mofetil
Mycophenolate mofetil is a recently introduced oral prepara-tion for use as an immunosuppressant in renal transplantation.
After absorption it is hydrolysed to
mycophenolic acid (MPA), which is an active metabolite, and is a potent
inhibitor of inosine monophosphate dehydrogenase which is necessary for the
synthesis of purines. Mycophenolate mofetil suppresses lymphocyte proliferation
and antibody formation by B cells.
Toxicity results in bone marrow
suppression and hepatic dysfunction.
Antibody Reagents
Antibody
reagents represent a promising therapeutic strategy, as they cause rapid
lowering of lymphocytes, as well as suppres-sion of function of specific
lymphocyte populations.
Antithymocyte globulin (Atgam)
It
is a purified immunoglobulin prepared commercially from hyperimmune serum of
horse, rabbit, sheep, or goat, following immunisation with human thymic
lymphocytes. It is used primarily to treat allograft rejection in kidney and
heart trans-plantation. Toxic effects include anaphylaxis, serum sickness,
nephritis, leukopenia, thrombocytopenia and fever.
Muromonab-CD3 monoclonal antibody
This
is a mouse monoclonal antibody which causes a more consistent immune
suppressive response than Atgam. It has been used to prevent acute rejection of
kidney, liver, and heart transplants. Adverse effects include anaphylactoid
reactions, cytokine release syndrome,*
and CNS toxicity.
Rh(D) immune globulin
This
antibody is prepared by alcohol fraction of plasma from donors, and is used in
Rh-negative mothers to prevent sensitisa-tion to Rh(D) antigen (to prevent
erythroblastosis foetalis). It is given intramuscularly. Adverse effects
include local pain, fever and anaphylaxis.
Related Topics