Chapter: Basic & Clinical Pharmacology : Antiviral Agents
Treatment of Hepatitis B Virus Infection
TREATMENT OF HEPATITIS B VIRUS
INFECTION
The
goals of chronic HBV therapy are to sustain suppression of HBV replication,
resulting in slowing of progression of hepatic disease (with retardation of
hepatic fibrosis and even reversal of cirrhosis), prevention of complications
(ie, cirrhosis, hepatic failure, and hepa-tocellular carcinoma), and reduction
of the need for liver transplanta-tion. This translates into suppression of HBV
DNA to undetectable levels, seroconversion of HBeAg (or more rarely, HBsAg)
from posi-tive to negative, and reduction in elevated hepatic transaminase
lev-els. These end points are correlated with improvement in necroinflammatory
disease, a decreased risk of hepatocellular carci-noma and cirrhosis, and a
decreased need for liver transplantation. All the currently licensed therapies
achieve these goals. However, because current therapies suppress HBV
replication without eradi-cating the virus, initial responses may not be
durable. The covalently closed circular (ccc) viral DNA exists in stable form
indefinitely within the cell, serving as a reservoir for HBV throughout the
life of the cell and resulting in the capacity to reactivate. Relapse is more
common in patients co-infected with HBV and hepatitis D virus.
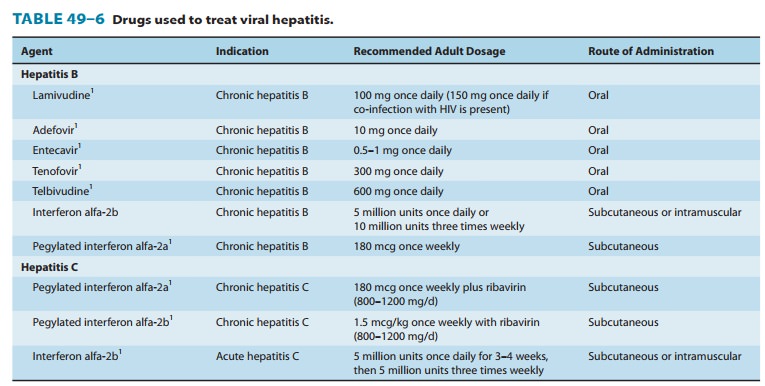
As of 2010 seven drugs
were approved for treatment of chronic HBV infection in the United States: five
oral nucleoside/nucleotide analogs (lamivudine, adefovir dipivoxil, tenofovir,
entecavir, telbivudine) and two injectable interferon drugs (interferon
alfa-2b, pegylated interferon alfa-2a) (Table 49–6). The use of interferon has
been supplanted by long-acting pegylated interferon, allowing once-weekly
rather than daily or thrice-weekly dosing. In general, nucleoside/nucleotide
analog therapies have better tolerability and ultimately produce a higher
response rate than the interferons (though less rapid); however, response may
be less sustained after discontinuation of the nucleoside/nucleotide therapies,
and emer-gence of resistance may be problematic. The nucleotides are effec-tive
in nucleoside resistance and vice versa. In addition, oral agents may be used
in patients with decompensated liver disease, and the therapy is chronic rather
than finite as with interferon therapy.
Several anti-HBV
agents have anti-HIV activity as well, includ-ing lamivudine, adefovir
dipivoxil, and tenofovir. Emtricitabine, an antiretroviral NRTI, is under clinical
evaluation for HBV treat-ment in combination with tenofovir, which may be
particularly suited to individuals co-infected with HIV and HBV. Because NRTI
agents may be used in patients co-infected with HBV and HIV, it is important to
note that acute exacerbation of hepatitis may occur upon discontinuation or
interruption of these agents.
ADEFOVIR DIPIVOXIL
Although initially and
abortively developed for treatment of HIV infection, adefovir dipivoxil gained
approval, at lower and less toxic doses, for treatment of HBV infection.
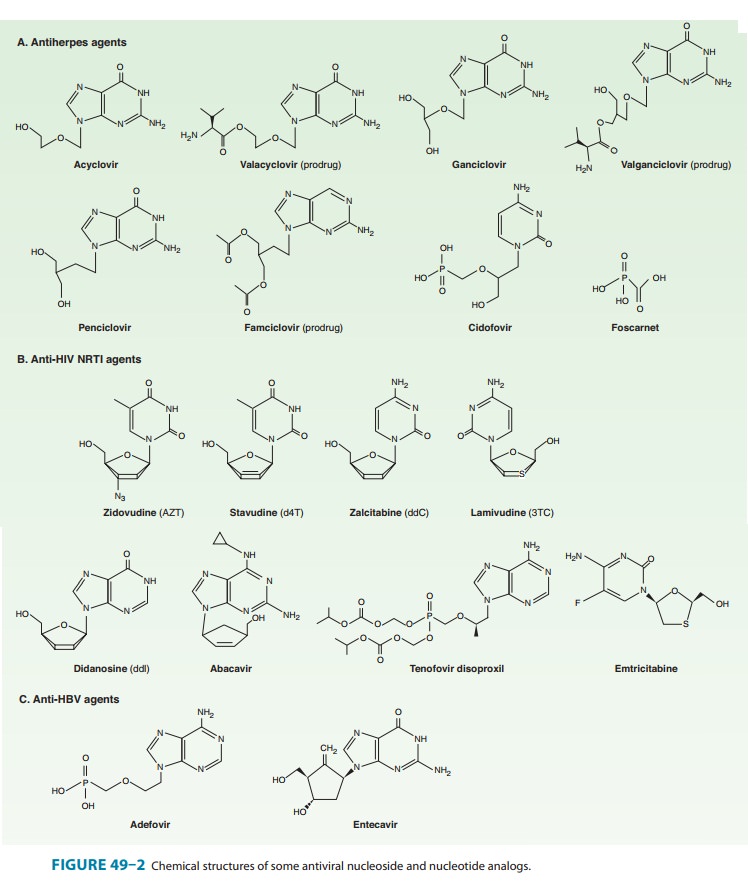
Adefovir dipivoxil is the diester prodrug of adefovir, an acyclic phosphonated
adenine nucleotide analog (Figure 49–2). It is phosphorylated by cellular
kinases to the active diphosphate metabolite and then competi-tively inhibits HBV
DNA polymerase and causes chain termina-tion after incorporation into viral
DNA. Adefovir is active in vitro against a wide range of DNA and RNA viruses,
including HBV, HIV, and herpesviruses.
Oral bioavailability
of adefovir dipivoxil is about 59% and is unaffected by meals; it is rapidly
and completely hydrolyzed to the parent compound by intestinal and blood
esterases. Protein bind-ing is low (< 5%). The intracellular half-life of the
diphosphate is prolonged, ranging from 5 to 18 hours in various cells; this
makes once-daily dosing feasible. Adefovir is excreted by a combination of
glomerular filtration and active tubular secretion and requires dose adjustment
for renal dysfunction; however, it may be admin-istered to patients with
decompensated liver disease.
Of
the oral agents, adefovir may be slower to suppress HBV DNA levels and the
least likely to induce HBeAg seroconversion. Although emergence of resistance
is rare during the first year of therapy, it approaches 30% at the end of 4
years. Naturally occur-ring (ie, primary) adefovir-resistant rt233 HBV mutants
have recently been described. There is no cross-resistance between adefovir and
lamivudine.
Adefovir dipivoxil is
well tolerated. A dose-dependent nephro-toxicity has been observed in clinical
trials, manifested by increased serum creatinine with decreased serum
phosphorous and more common in patients with baseline renal insufficiency and
those receiving high doses (60 mg/d). Other potential adverse effects are
headache, diarrhea, asthenia, and abdominal pain. As with other NRTI agents,
lactic acidosis and hepatic steatosis are considered a risk owing to
mitochondrial dysfunction. No clini-cally important drug-drug interactions have
been recognized todate; however, co-administration with drugs that reduce renal
function or compete for active tubular secretion may increase serum
concentrations of adefovir or the co-administered drug. Pivalic acid, a
by-product of adefovir dipivoxil metabolism, can esterify free carnitine and
result in decreased carnitine levels. However, it is not felt necessary to
administer carnitine supple-mentation with the low doses used to treat patients
with HBV (10 mg/d). Adefovir is embryotoxic in rats at high doses and is
genotoxic in preclinical studies.
ENTECAVIR
Entecavir is an orally
administered guanosine nucleoside analog (Figure 49–2) that competitively
inhibits all three functions of HBV DNA polymerase, including base priming,
reverse transcrip-tion of the negative strand, and synthesis of the positive
strand of HBV DNA. Oral bioavailability approaches 100% but is decreased by
food; therefore, entecavir should be taken on an empty stomach. The
intracellular half-life of the active phosphorylated compound is 15 hours. It
is excreted by the kidney, undergoing both glomerular filtration and net
tubular secretion.
Comparison with
lamivudine in patients with chronic HBV infection demonstrated similar rates of
HBeAg seroconversion but significantly higher rates of HBV DNA viral
suppression with entecavir, normalization of serum alanine aminotransferase
levels, and histologic improvement in the liver. Entecavir appears to have a
higher barrier to the emergence of resistance than lamivudine. Although
selection of resistant isolates with the S202G mutation has been documented
during therapy, clinical resistance is rare (< 1% at 4 years). Also,
decreased susceptibility to entecavir may occur in association with lamivudine
resistance.
Entecavir is well
tolerated. The most frequent adverse events are headache, fatigue, dizziness,
and nausea. Lung adenomas and carci-nomas in mice, hepatic adenomas and
carcinomas in rats and mice, vascular tumors in mice, and brain gliomas and
skin fibromas in rats have been observed at varying exposures.
Co-administration of entecavir with drugs that reduce renal function or compete
for active tubular secretion may increase serum concentrations of either
entecavir or the co-administered drug.
LAMIVUDINE
The pharmacokinetics
of lamivudine are described earlier (see section, Nucleoside and Nucleotide
Reverse Transcriptase Inhibitors). The more prolonged intracellular half-life
in HBV cell lines (17–19 hours) than in HIV-infected cell lines (10.5–15.5
hours) allows for lower doses and less frequent administration. Lamivudine can
be safely administered to patients with decompensated liver disease.
Lamivudine inhibits
HBV DNA polymerase and HIV reverse transcriptase by competing with
deoxycytidine triphosphate for incorporation into the viral DNA, resulting in
chain termination. Lamivudine achieves 3–4 log decreases in viral replication
in most patients and suppression of HBV DNA to undetectable levels in about 44%
of patients. Seroconversion of HBeAg from positive to negative occurs in about
17% of patients and is durable at 3 years in about 70% of responders.
Continuation of treatment for 4–8 months after seroconversion may improve the
durability of response. Response in HBeAg-negative patients is initially high
but less durable.
Although
lamivudine initially results in rapid and potent virus suppression, chronic
therapy may ultimately be limited by the emer-gence of lamivudine-resistant HBV
isolates (eg, L180M or M204I/V), estimated to occur in 15–30% of patients at 1
year and 70% at 5 years of therapy. Resistance has been associated with flares
of hepati-tis and progressive liver disease. Cross-resistance between
lamivudine and emtricitabine or entecavir may occur; however, adefovir
main-tains activity against lamivudine-resistant strains of HBV.
In the doses used for
HBV infection, lamivudine has an excel-lent safety profile. Headache, nausea,
and dizziness are rare. Co-infection with HIV may increase the risk of
pancreatitis. No evidence of mitochondrial toxicity has been reported.
TELBIVUDINE
Telbivudine is a
thymidine nucleoside analog with activity against HBV DNA polymerase. It is
phosphorylated by cellular kinases to the active triphosphate form, which has
an intracellular half-life of 14 hours. The phosphorylated compound
competitively inhibits HBV DNA polymerase, resulting in incorporation into
viral DNA and chain termination. It is not active in vitro against HIV-1.
Oral bioavailability
is unaffected by food. Plasma protein-binding is low (3%) and distribution
wide. The serum half-life is approximately 15 hours and excretion is renal.
There are no known metabolites and no known interactions with the CYP450 system
or other drugs.
In
a comparative trial against lamivudine in patients with chronic HBV infection,
significantly more patients receiving telbivudine achieved the combined end
point of suppression of HBV DNA to less than 5 log copies/mL plus loss of serum
HBeAg. The mean reduction in HBV DNA from baseline, the proportion with ALT
normalization, and HBeAg seroconversion all were greater in those receiving
telbivudine. Liver biopsies per-formed 1 year later showed less scarring.
However, emergence of resistance, typically due to the M204I mutation, may
occur in up to 22% of patients with durations of therapy exceeding 1 year, and
may result in virologic rebound.
Adverse
effects are mild; they include fatigue, headache, abdominal pain, upper
respiratory infection, increased creatine kinase levels, and nausea and
vomiting. Both uncomplicated myalgia and myopathy have been reported, as has
peripheral neuropathy. As with other nucleoside analogs, lactic acidosis and
severe hepatomegaly with steatosis may occur during therapy as well as flares
of hepatitis after discontinuation.
TENOFOVIR
Tenofovir, a
nucleotide analog of adenosine in use as an antiretro-viral agent, has recently
gained licensure for the treatment ofpatients with chronic HBV infection. The
characteristics of teno-fovir are described earlier. Tenofovir maintains
activity against lamivudine- and entecavir-resistant hepatitis virus isolates
but has reduced activity against adefovir-resistant strains. Although similar
in structure to adefovir dipivoxil, comparative trials showed a significantly
higher rate of complete response, defined as serum HBV DNA levels less than 400
copies/mL, as well as of histologic improvement, in patients with chronic HBV
infection receiving tenofovir than in those receiving adefovir dipivoxil. The
emergence of resistance appears to be substantially less frequent during
therapy with tenofovir than with adefovir.
INVESTIGATIONAL AGENTS
Compounds in clinical
development for the treatment of patients with HBV infection include the
nucleoside analogs emtricitabine and
clevudine, as well as the
immunologic modulator thymosinalpha-1, agents
that facilitate uptake by the liver using conjuga-tion to ligands, and RNA
interference compounds.
Related Topics