Chapter: Basic & Clinical Pharmacology : Antiviral Agents
Nonnucleoside Reverse Transcriptase Inhibitors
NONNUCLEOSIDE REVERSE
TRANSCRIPTASE INHIBITORS
The NNRTIs bind
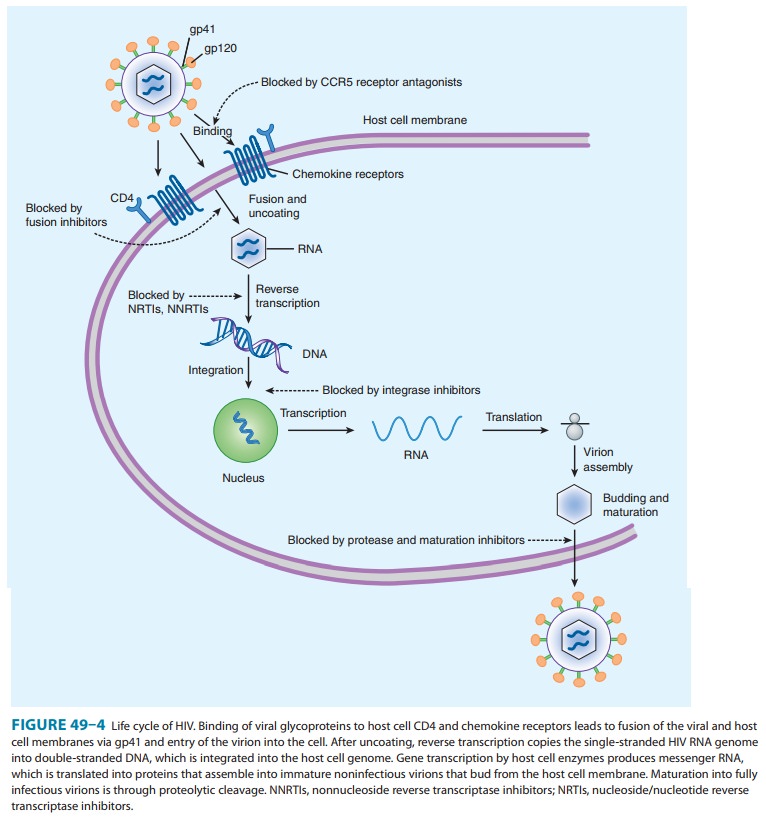
directly to HIV-1 reverse transcriptase (Figure 49–4), resulting in allosteric
inhibition of RNA- and DNA-dependent DNA polymerase activity. The binding site
of NNRTIs is near to but distinct from that of NRTIs. Unlike the NRTI agents,
NNRTIs neither compete with nucleoside triphosphates nor require
phosphorylation to be active.
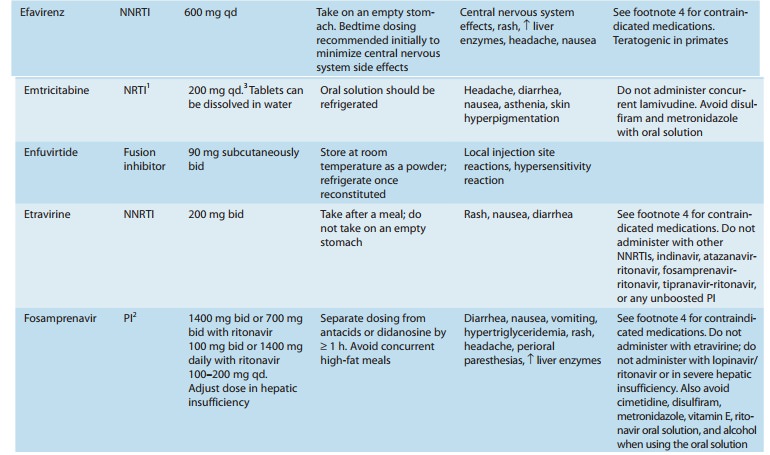
Baseline genotypic
testing is recommended prior to initiating NNRTI treatment because primary
resistance rates range from approximately 2% to 8%. NNRTI resistance occurs
rapidly with monotherapy and can result from a single mutation. The K103N and
Y181C mutations confer resistance across the entire class of NNRTIs, with the
exception of the newest agent, etravirine. Other mutations (eg, L100I, Y188C,
G190A) may confer cross-resistance among the NNRTI class. However, there is no
cross-resistance between the NNRTIs and the NRTIs; in fact, some
nucleoside-resistant viruses display hypersusceptibility to NNRTIs.
As
a class, NNRTI agents tend to be associated with varying levels of
gastrointestinal intolerance and skin rash, the latter of which may
infrequently be serious (eg, Stevens-Johnson syndrome). A further limitation to
use of NNRTI agents as a component of antiretroviral therapy is their
metabolism by the CYP450 system, leading to innumerable potential drug-drug
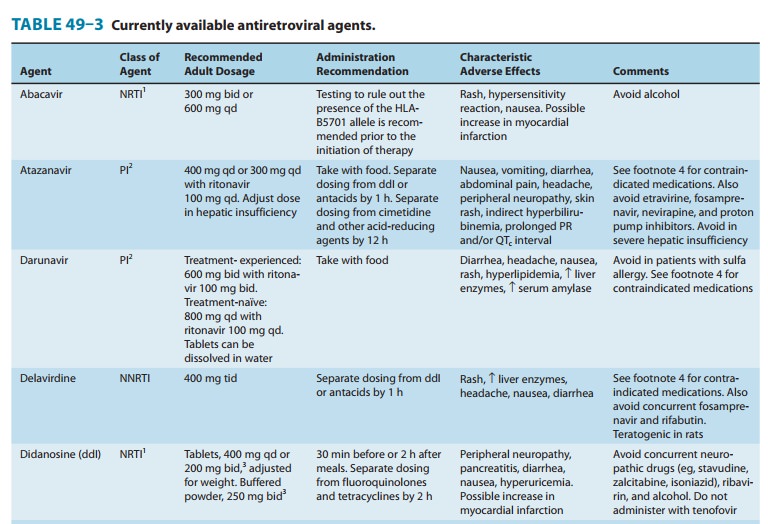
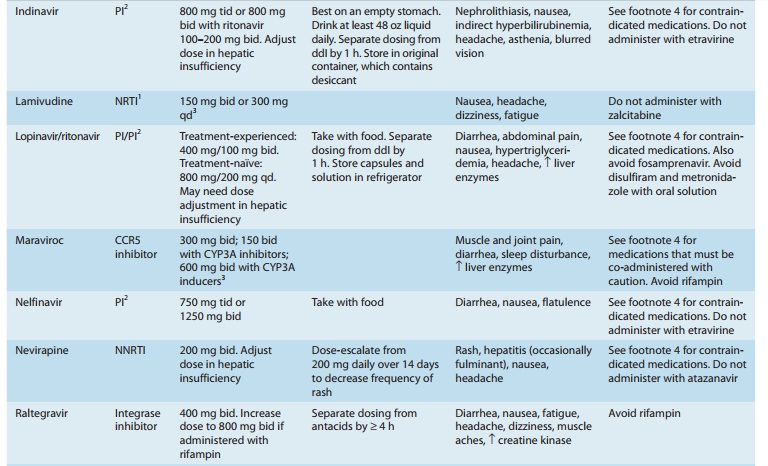
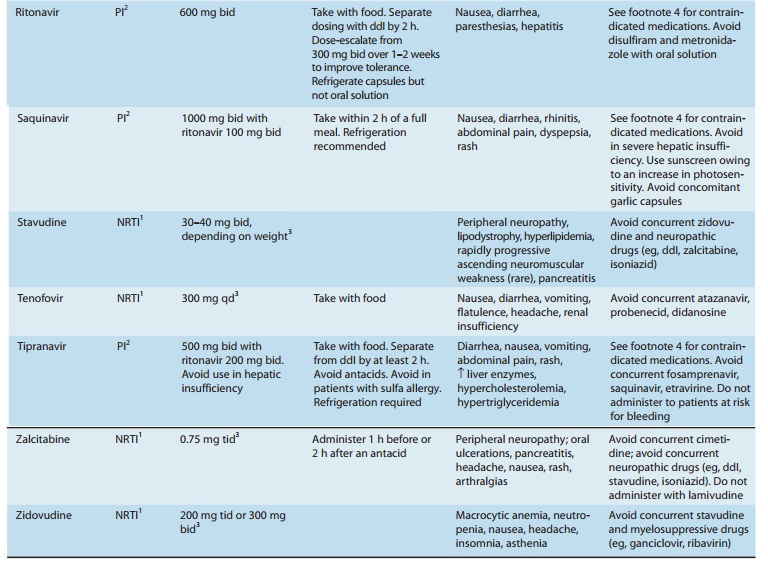
interactions (Tables 49–3 and 49–4). All NNRTI agents are substrates for CYP3A4
and can act as inducers (nevirapine), inhibitors (delavirdine), or mixed
inducers and inhibitors (efavirenz, etravirine). Given the large number of
non-HIV medications that are also metabolized by this pathway , drug-drug interactions
must be expected and looked for; dosage adjustments are frequently required and
some combinations are contraindicated.
DELAVIRDINE
Delavirdine has an
oral bioavailability of about 85%, but this is reduced by antacids or H 2-blockers. It is
extensively bound (∼ 98%) to plasma
proteins and has correspondingly low cerebrospi-nal fluid levels. Serum
half-life is approximately 6 hours.
Skin rash occurs in up
to 38% of patients receiving delavirdine; it typically occurs during the first
1–3 weeks of therapy and does not preclude rechallenge. However, severe rash
such as erythema multiforme and Stevens-Johnson syndrome have rarely been
reported. Other possible adverse effects are headache, fatigue, nausea,
diarrhea, and increased serum aminotransferase levels.
Delavirdine has been
shown to be teratogenic in rats, causing ventricular septal defects and other
malformations at dosages not unlike those achieved in humans. Thus, pregnancy
should be avoided when taking delavirdine.
Delavirdine
is extensively metabolized by the CYP3A and CYP2D6 enzymes and also inhibits
CYP3A4 and 2C9. Therefore, there are numerous potential drug-drug interactions
to consider (Tables 49–3 and 49–4). The concurrent use of delavirdine with
fosamprenavir and rifabutin is not recom-mended because of decreased
delavirdine levels. Other medica-tions likely to alter delavirdine levels
include didanosine, lopinavir, nelfinavir, and ritonavir. Co-administration of
dela-virdine with indinavir or saquinavir prolongs the elimination half-life of
these protease inhibitors, thus allowing them to be dosed twice rather than
thrice daily.
EFAVIRENZ
Efavirenz can be given
once daily because of its long half-life (40–55 hours). It is moderately well
absorbed following oral administration (45%). Since toxicity may increase owing
to increased bioavailability after a high-fat meal, efavirenz should be taken
on an empty stomach. Efavirenz is principally metabolized by CYP3A4 and CYP2B6
to inactive hydroxylated metabolites; the remainder is eliminated in the feces
as unchanged drug. It is highly bound to albumin (∼ 99%), and cerebrospinal fluid levels range
from 0.3% to 1.2% of plasma levels.
The principal adverse
effects of efavirenz involve the central nervous system. Dizziness, drowsiness,
insomnia, nightmares, and headache tend to diminish with continued therapy;
dosing at bedtime may also be helpful. Psychiatric symptoms such as
depres-sion, mania, and psychosis have been observed and may necessi-tate
discontinuation. Skin rash has also been reported early in therapy in up to 28%
of patients; the rash is usually mild to moderate in severity and typically
resolves despite continuation. Rarely, rash has been severe or
life-threatening. Other potential adverse reactions are nausea, vomiting,
diarrhea, crystalluria, ele-vated liver enzymes, and an increase in total serum
cholesterol by 10–20%. High rates of fetal abnormalities occurred in pregnant
monkeys exposed to efavirenz in doses roughly equivalent to the human dosage;
several cases of congenital anomalies have been reported in humans. Therefore,
efavirenz should be avoided in pregnant women, particularly in the first
trimester.
As
both an inducer and an inhibitor of CYP3A4, efavirenz induces its own
metabolism and interacts with the metabolism of many other drugs (Tables 49–3
and 49–4). Since efavirenz may lower methadone levels, patients receiving these
two agents con-currently should be monitored for signs of opioid withdrawal and
may require an increased dose of methadone.
ETRAVIRINE
In
2008, etravirine was approved in the United States for use in
treatment-experienced patients with HIV infection. Etravirinemay be effective
against strains of HIV that have developed resis-tance to first-generation
NNRTIs, depending on the number of mutations present. Although etravirine has a
higher genetic barrier to resistance than the other NNRTIs, mutations selected
by etravirine usually are associated with resistance to efavirenz, nevirapine,
and delavirdine.
The most common
adverse effects of etravirine are rash, nausea, and diarrhea. The rash is
typically mild and usually resolves after 1–2 weeks without discontinuation of
therapy. Rarely, rash has been severe or life-threatening. Laboratory
abnormalities include elevations in serum cholesterol, triglyceride, glucose,
and hepatic transaminase levels. Transaminase elevations are more common in
patients with HBV or HCV co-infection.
Etravirine
is a substrate as well as an inducer of CYP3A4 and an inhibitor of CYP2C9 and
CYP2C19; it has many therapeuti-cally significant drug-drug interactions
(Tables 49–3 and 49–4). Some of the interactions are difficult to predict. For
example, etravirine may decrease itraconazole and ketoconazole concentra-tions
but increase voriconazole concentrations.
NEVIRAPINE
The oral bioavailability
of nevirapine is excellent (> 90%) and is not food-dependent. The drug is highly lipophilic
and achieves cerebrospinal fluid levels that are 45% of those in plasma. Serum
half-life is 25–30 hours. It is extensively metabolized by the CYP3A isoform to
hydroxylated metabolites and then excreted, primarily in the urine.
A
single dose of nevirapine (200 mg) is effective in the preven-tion of
transmission of HIV from mother to newborn when administered to women at the
onset of labor and followed by a 2 mg/kg oral dose to the neonate within 3 days
after delivery. There is no evidence of human teratogenicity. However,
resistance has been documented after this single dose.
Rash, usually a
maculopapular eruption that spares the palms and soles, occurs in up to 20% of
patients, usually in the first 4–6 weeks of therapy. Although typically mild
and self-limited, rash is dose-limiting in about 7% of patients. Women appear
to have an increased incidence of rash. When initiating therapy, gradual dose
escalation over 14 days is recommended to decrease the incidence of rash.
Severe and life-threatening skin rashes have been rarely reported, including
Stevens-Johnson syndrome and toxic epidermal necrolysis. Nevirapine therapy
should be immedi-ately discontinued in patients with severe rash and in those
with accompanying constitutional symptoms; since rash may accom-pany
hepatotoxicity, liver function tests should be assessed. Symptomatic liver
toxicity may occur in up to 4% of patients and is more frequent in those with higher
pretherapy CD4 cell counts (ie, > 250 cells/mm3 in women and > 400 cells/mm3 in men), in women, and in those with HBV or HCV co-infection.
Fulminant, life-threatening hepatitis has been reported, typically within the
first 18 weeks of therapy. Other adverse effects include fever, nau-sea,
headache, and somnolence.
Nevirapine
is a moderate inducer of CYP3A metabolism, resulting in decreased levels of
amprenavir, indinavir, lopinavir,saquinavir, efavirenz, and methadone. Drugs
that induce the CYP3A system, such as rifampin, rifabutin, and St. John’s wort,
can decrease levels of nevirapine, whereas those that inhibit CYP3A activity,
such as fluconazole, ketoconazole, and clarithro-mycin, can increase nevirapine
levels. Since nevirapine may lower methadone levels, patients receiving these
two agents concurrently should be monitored for signs of opioid withdrawal and
may require an increased dose of methadone.
Related Topics