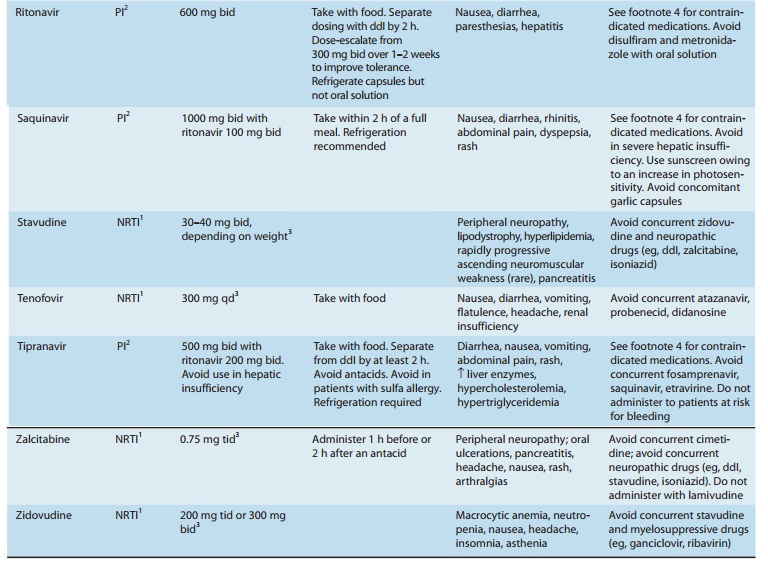
Chapter: Basic & Clinical Pharmacology : Antiviral Agents
Protease Inhibitors
PROTEASE INHIBITORS
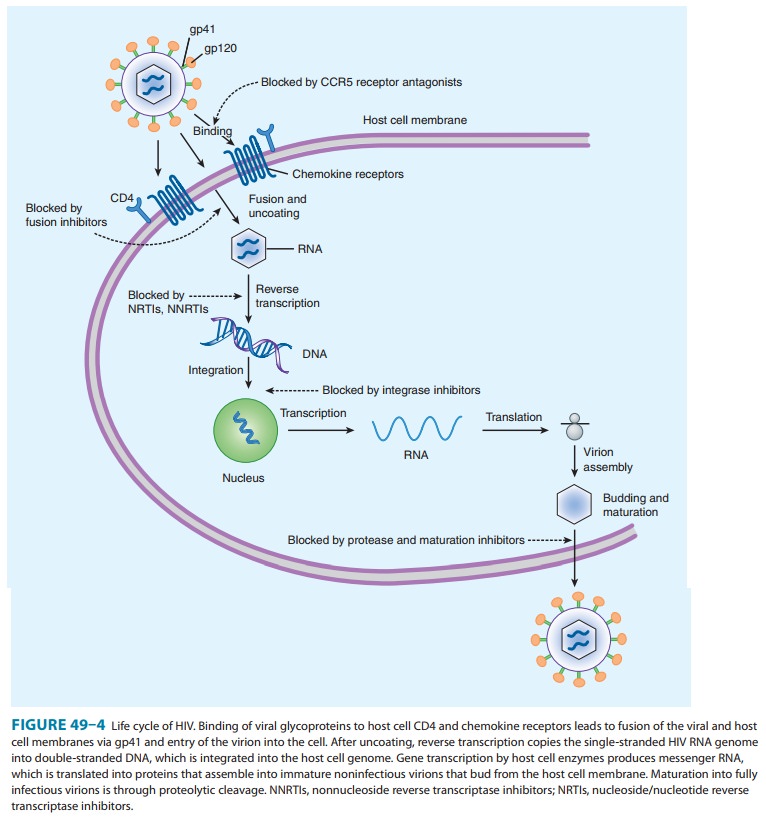
During the later
stages of the HIV growth cycle, the gag
and gag-pol gene products are translated into polyproteins, and thesebecome
immature budding particles. The HIV protease is respon-sible for cleaving these
precursor molecules to produce the final structural proteins of the mature
virion core. By preventing post-translational cleavage of the Gag-Pol
polyprotein, protease inhibi-tors (PIs) prevent the processing of viral
proteins into functional conformations, resulting in the production of
immature, nonin-fectious viral particles (Figure 49–4). Unlike the NRTIs, PIs
do not need intracellular activation.
Specific genotypic
alterations that confer phenotypic resistance are fairly common with these
agents, thus contraindicating mono-therapy. Some of the most common mutations
conferring broad resistance to PIs are substitutions at the 10, 46, 54, 82, 84,
and 90 codons; the number of mutations may predict the level of phe-notypic
resistance. The I50L substitution emerging during atazana-vir therapy has been
associated with increased
susceptibility to other PIs. Darunavir and tipranavir appear to have improved
virologic activity in patients harboring HIV-1 resistant to other PIs.
A
syndrome of redistribution and accumulation of body fat that results in central
obesity, dorsocervical fat enlargement (buffalo hump), peripheral and facial
wasting, breast enlargement, and a cushingoid appearance has been observed in
patients receiv-ing antiretroviral therapy. These abnormalities may be
particularly associated with the use of PIs, although the recently licensed
atazanavir appears to be an exception . Concurrent increases in triglyceride
and low-density lipoprotein levels, along with hyperglycemia and insulin
resistance, have also been noted. The cause is not yet known.
Whether
PI agents are associated with bone loss and osteoporosis after long-term use is
controversial and under investigation. PIs have been associated with increased
spontaneous bleeding in patients with hemophilia A or B; an increased risk of
intracranial hemorrhage has been reported in patients receiving tipranavir with
ritonavir.
The
concurrent use of saquinavir and ritonavir has recently been found to be
associated with QT and PR interval prolonga-tion, and is contraindicated. QT
prolongation may result in life-threatening torsades de pointes arrhythmia.
All of the
antiretroviral PIs are extensively metabolized by CYP3A4, with ritonavir having
the most pronounced inhibitory effect and saquinavir the least. Some PI agents,
such as amprenavir and ritonavir, are also inducers of specific CYP isoforms.
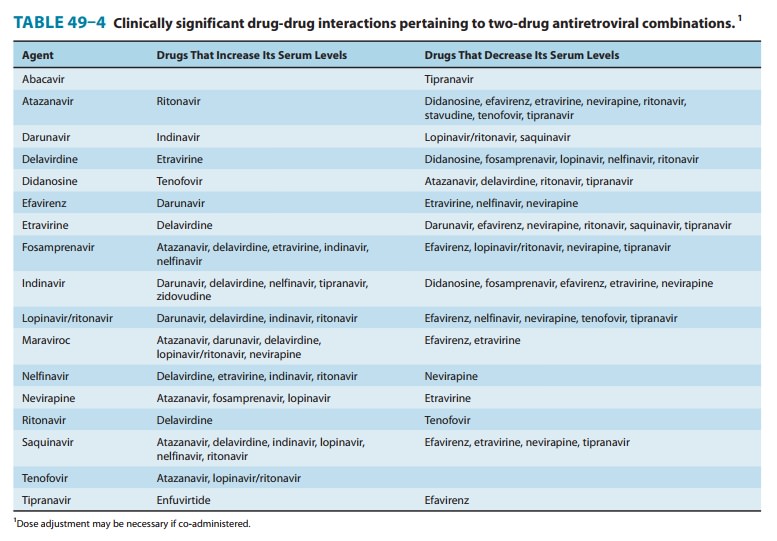
As a result, there is enormous potential for drug-drug interactions with other
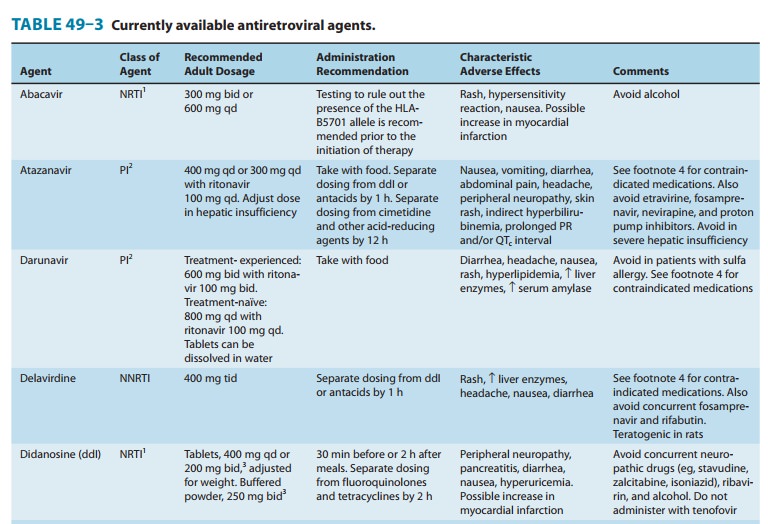
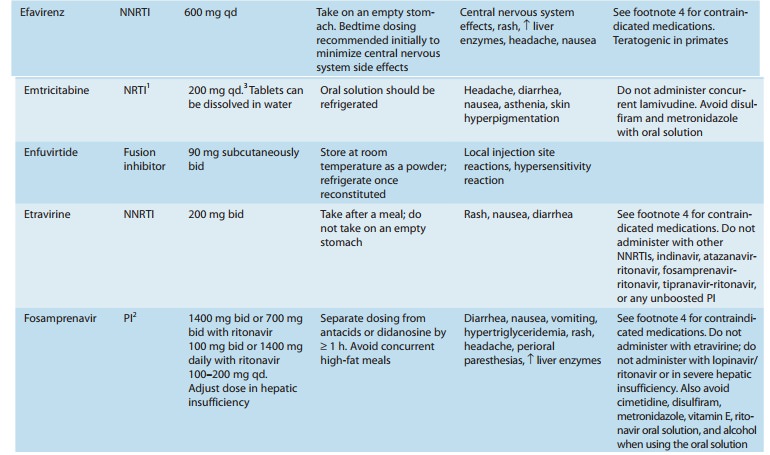
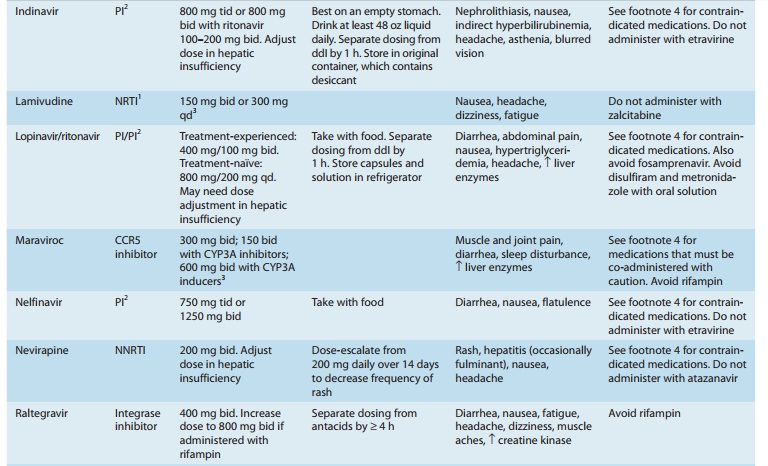
antiretroviral agents and other commonly used medications(Tables 49–3 and
49–4). Expert resources about drug-drug inter-actions should be consulted, as
dosage adjustments are frequently required and some combinations are
contraindicated. It is note-worthy that the potent CYP3A4 inhibitory properties
of ritonavir are used to clinical advantage by having it “boost” the levels of
other PI agents when given in combination, thus acting as a phar-macokinetic
enhancer rather than an antiretroviral agent. Ritonavir boosting increases drug
exposure, thereby prolonging the drug’s half-life and allowing reduction in
frequency; in addition, the genetic barrier to resistance is raised.
ATAZANAVIR
Atazanavir
is an azapeptide PI with a pharmacokinetic profile that allows once-daily
dosing. It should be taken with a light meal to enhance bioavailability.
Atazanavir requires an acidic medium for absorption and exhibits pH-dependent
aqueous solubility; there-fore, separation of ingestion from acid-reducing
agents by at least 12 hours is recommended and concurrent proton pump
inhibitors are contraindicated. Atazanavir is able to penetrate both the
cerebro-spinal and seminal fluids. The plasma half-life is 6–7 hours, which
increases to approximately 11 hours when co-administered with ritonavir. The
primary route of elimination is biliary; atazanavir should not be given to
patients with severe hepatic insufficiency.
Resistance to
atazanavir has been associated with various known PI mutations as well as with
the novel I50L substitution. Whereas some atazanavir resistance mutations have
been associated in vitro with decreased susceptibility to other PIs, the I50L
mutation has been associated with increased susceptibility to other PIs.
The most common
adverse effects in patients receiving ataza-navir are diarrhea and nausea;
vomiting, abdominal pain, head-ache, peripheral neuropathy, and skin rash may
also occur. As with indinavir, indirect hyperbilirubinemia with overt jaundice
may occur in approximately 10% of patients, owing to inhibition of the UGT1A1
glucuronidation enzyme. Elevation of hepatic enzymes has also been observed,
usually in patients with underly-ing HBV or HCV co-infection. Nephrolithiasis
has recently been described in association with atazanavir use. In contrast to
the other PIs, atazanavir does not appear to be associated with dys-lipidemia,
fat redistribution, or the metabolic syndrome. Atazanavir may be associated
with prolongation of the electrocardiographic PR interval, which is usually
inconsequential but may be exacer-bated by other causative agents such as
calcium channel blockers and may result in AV block QT prolongation is another
potential electrocardiographic effect of atazanavir but is rarely clinically
significant.
As an inhibitor of
CYP3A4 and CYP2C9, the potential for drug-drug interactions with atazanavir is
great (Tables 49–3 and 49–4). Atazanavir AUC is reduced by up to 76% when
combined with a proton pump inhibitor; thus, these combinations are to be
avoided. In addition, co-administration of atazanavir with other drugs that
inhibit UGT1A1, such as irinotecan, may increase its levels. Tenofovir and
efavirenz should not be co-administered with atazanavir unless ritonavir is
added to boost levels.
DARUNAVIR
Darunavir
is licensed as a PI that must be co-administered with ritonavir. It was
initially licensed for use in treatment-experienced patients only; thus there
is less clinical experience with its use in treatment-naïve patients. Darunavir
may be administered once daily in treatment-naïve patients.Symptomatic adverse
effects of darunavir include diarrhea, nausea, headache, and rash. Laboratory
abnormalities include dyslipidemia (though possibly less frequent than with
other boosted PI regimens) and increases in amylase and hepatic transaminase
levels. Liver toxicity, including severe hepatitis, has been reported in some
patients taking darunavir; the risk of hepatotoxicity may be higher for persons
with HBV, HCV, or other chronic liver disease.Darunavir contains a sulfonamide
moiety and should be used cautiously in patients with sulfonamide
allergy.Darunavir both inhibits and is metabolized by the CYP3A enzyme system,
conferring many possible drug-drug interactions (Tables 49–3 and 49–4). In
addition, the co-administered ritona-vir is a potent inhibitor of CYP3A and
CYP2D6, and an inducer of other hepatic enzyme systems.
FOSAMPRENAVIR
Fosamprenavir
is a prodrug of amprenavir that is rapidly hydro-lyzed by enzymes in the
intestinal epithelium. Because of its sig-nificantly lower daily pill burden,
fosamprenavir tablets have replaced amprenavir capsules for adults.
Fosamprenavir is most often administered in combination with low-dose
ritonavir.
Amprenavir
is rapidly absorbed from the gastrointestinal tract, and its prodrug can be
taken with or without food. However, high-fat meals decrease absorption and
thus should be avoided. The plasma half-life is relatively long (7–11 hours).
Amprenavir is metabolized in the liver by CYP3A4 and should be used with
caution in the setting of hepatic insufficiency.
The
most common adverse effects of fosamprenavir are headache, nausea, diarrhea,
perioral paresthesias, depression, and rash. Up to 3% of patients may
experience rashes (including Stevens-Johnson syndrome) severe enough to warrant
drug discontinuation.
Amprenavir is both an inducer and an inhibitor of CYP3A4 and
is contraindicated with numerous drugs (Tables 49–3 and 49–4). The oral
solution, which contains propylene glycol, is con-traindicated in young
children, pregnant women, patients with renal or hepatic failure, and those
using metronidazole or disul-firam. Also, the oral solutions of amprenavir and
ritonavir should not be co-administered because the propylene glycol in one and
the ethanol in the other may compete for the same metabolic pathway, leading to
accumulation of either. Because the oral solu-tion also contains vitamin E at
several times the recommended daily dosage, supplemental vitamin E should be
avoided. Amprenavir, a sulfonamide, is contraindicated in patients with a
history of sulfa allergy. Lopinavir/ritonavir should not be co-administered
with amprenavir owing to decreased amprenavir andaltered lopinavir exposures.
An increased dosage of amprenavir is recommended when co-administered with
efavirenz (with or without the addition of ritonavir to boost levels).
INDINAVIR
Indinavir requires an
acidic environment for optimum solubility and therefore must be consumed on an
empty stomach or with a small, low-fat, low-protein meal for maximal absorption
(60–65%). The serum half-life is 1.5–2 hours, protein binding is approxi-mately
60%, and the drug has a high level of cerebrospinal fluid penetration (up to
76% of serum levels). Excretion is primarily fecal. An increase in AUC by 60%
and in half-life to 2.8 hours in the setting of hepatic insufficiency
necessitates dose reduction.
The most common
adverse effects of indinavir are indirect hyperbilirubinemia and
nephrolithiasis due to urinary crystalliza-tion of the drug. Nephrolithiasis
can occur within days after initiat-ing therapy, with an estimated incidence of
approximately 10%. Consumption of at least 48 ounces of water daily is
important to maintain adequate hydration. Thrombocytopenia, elevations of serum
aminotransferase levels, nausea, diarrhea, insomnia, dry throat, dry skin, and
indirect hyperbilirubinemia have also been reported. Insulin resistance may be
more common with indinavir than with the other PIs, occurring in 3–5% of
patients. There have also been rare cases of acute hemolytic anemia. In rats,
high doses of indinavir are associated with development of thyroid adenomas.
Since indinavir is an
inhibitor of CYP3A4, numerous and complex drug interactions can occur (Tables
49–3 and 49–4). Combination with ritonavir (boosting) allows for twice-daily
rather than thrice-daily dosing and eliminates the food restriction associated
with use of indinavir. However, there is potential for an increase in
nephrolithiasis with this combination compared with indinavir alone; thus, a
high fluid intake (1.5–2 L/d) is advised.
LOPINAVIR
Lopinavir
is currently formulated only in combination with ritonavir, which inhibits the
CYP3A-mediated metabolism of lopinavir, thereby resulting in increased exposure
to lopinavir. In addition to improved patient compliance due to reduced pill
burden, lopinavir/ritonavir is generally well tolerated.Lopinavir should be
taken with food to enhance bioavailability. The drug is highly protein bound
(98–99%), and its half-life is 5–6 hours. Lopinavir is extensively metabolized
by CYP3A, which is inhibited by ritonavir. Serum levels of lopinavir may be
increased in patients with hepatic impairment.
The
most common adverse effects of lopinavir are diarrhea, abdominal pain, nausea,
vomiting, and asthenia. Elevations in serum cholesterol and triglycerides are
common. Potential drug-drug inter-actions are extensive (Tables 49–3 and 49–4).
Increased dosage of lopinavir/ritonavir is recommended when co-administered
with efavirenz or nevirapine, which induce lopinavir metabolism. Concurrent use
of fosamprenavir should be avoided owing to altered
DARUNAVIR
Darunavir
is licensed as a PI that must be co-administered with ritonavir. It was
initially licensed for use in treatment-experienced patients only; thus there
is less clinical experience with its use in treatment-naïve patients. Darunavir
may be administered once daily in treatment-naïve patients.Symptomatic adverse
effects of darunavir include diarrhea, nausea, headache, and rash. Laboratory
abnormalities include dyslipidemia (though possibly less frequent than with
other boosted PI regimens) and increases in amylase and hepatic transaminase
levels. Liver toxicity, including severe hepatitis, has been reported in some
patients taking darunavir; the risk of hepatotoxicity may be higher for persons
with HBV, HCV, or other chronic liver disease.
Darunavir contains a
sulfonamide moiety and should be used cautiously in patients with sulfonamide
allergy.
Darunavir both
inhibits and is metabolized by the CYP3A enzyme system, conferring many
possible drug-drug interactions (Tables 49–3 and 49–4). In addition, the
co-administered ritona-vir is a potent inhibitor of CYP3A and CYP2D6, and an
inducer of other hepatic enzyme systems.
FOSAMPRENAVIR
Fosamprenavir
is a prodrug of amprenavir that is rapidly hydro-lyzed by enzymes in the
intestinal epithelium. Because of its sig-nificantly lower daily pill burden,
fosamprenavir tablets have replaced amprenavir capsules for adults.
Fosamprenavir is most often administered in combination with low-dose
ritonavir.
Amprenavir
is rapidly absorbed from the gastrointestinal tract, and its prodrug can be
taken with or without food. However, high-fat meals decrease absorption and
thus should be avoided. The plasma half-life is relatively long (7–11 hours).
Amprenavir is metabolized in the liver by CYP3A4 and should be used with
caution in the setting of hepatic insufficiency.
The
most common adverse effects of fosamprenavir are headache, nausea, diarrhea,
perioral paresthesias, depression, and rash. Up to 3% of patients may
experience rashes (including Stevens-Johnson syndrome) severe enough to warrant
drug discontinuation.
Amprenavir is both an
inducer and an inhibitor of CYP3A4 and is contraindicated with numerous drugs
(Tables 49–3 and 49–4). The oral solution, which contains propylene glycol, is
con-traindicated in young children, pregnant women, patients with renal or
hepatic failure, and those using metronidazole or disul-firam. Also, the oral
solutions of amprenavir and ritonavir should not be co-administered because the
propylene glycol in one and the ethanol in the other may compete for the same
metabolic pathway, leading to accumulation of either. Because the oral
solu-tion also contains vitamin E at several times the recommended daily
dosage, supplemental vitamin E should be avoided. Amprenavir, a sulfonamide, is
contraindicated in patients with a history of sulfa allergy.
Lopinavir/ritonavir should not be co-administered with amprenavir owing to
decreased amprenavir andaltered lopinavir exposures. An increased dosage of
amprenavir is recommended when co-administered with efavirenz (with or without
the addition of ritonavir to boost levels).
INDINAVIR
Indinavir requires an
acidic environment for optimum solubility and therefore must be consumed on an
empty stomach or with a small, low-fat, low-protein meal for maximal absorption
(60–65%). The serum half-life is 1.5–2 hours, protein binding is approxi-mately
60%, and the drug has a high level of cerebrospinal fluid penetration (up to
76% of serum levels). Excretion is primarily fecal. An increase in AUC by 60%
and in half-life to 2.8 hours in the setting of hepatic insufficiency
necessitates dose reduction.
The most common
adverse effects of indinavir are indirect hyperbilirubinemia and
nephrolithiasis due to urinary crystalliza-tion of the drug. Nephrolithiasis
can occur within days after initiat-ing therapy, with an estimated incidence of
approximately 10%. Consumption of at least 48 ounces of water daily is
important to maintain adequate hydration. Thrombocytopenia, elevations of serum
aminotransferase levels, nausea, diarrhea, insomnia, dry throat, dry skin, and
indirect hyperbilirubinemia have also been reported. Insulin resistance may be
more common with indinavir than with the other PIs, occurring in 3–5% of
patients. There have also been rare cases of acute hemolytic anemia. In rats,
high doses of indinavir are associated with development of thyroid adenomas.
Since indinavir is an
inhibitor of CYP3A4, numerous and complex drug interactions can occur (Tables
49–3 and 49–4). Combination with ritonavir (boosting) allows for twice-daily
rather than thrice-daily dosing and eliminates the food restriction associated
with use of indinavir. However, there is potential for an increase in
nephrolithiasis with this combination compared with indinavir alone; thus, a high
fluid intake (1.5–2 L/d) is advised.
LOPINAVIR
Lopinavir
is currently formulated only in combination with ritonavir, which inhibits the
CYP3A-mediated metabolism of lopinavir, thereby resulting in increased exposure
to lopinavir. In addition to improved patient compliance due to reduced pill
burden, lopinavir/ritonavir is generally well tolerated.
Lopinavir
should be taken with food to enhance bioavailability. The drug is highly
protein bound (98–99%), and its half-life is 5–6 hours. Lopinavir is extensively
metabolized by CYP3A, which is inhibited by ritonavir. Serum levels of
lopinavir may be increased in patients with hepatic impairment.
The
most common adverse effects of lopinavir are diarrhea, abdominal pain, nausea,
vomiting, and asthenia. Elevations in serum cholesterol and triglycerides are
common. Potential drug-drug inter-actions are extensive (Tables 49–3 and 49–4).
Increased dosage of lopinavir/ritonavir is recommended when co-administered
with efavirenz or nevirapine, which induce lopinavir metabolism. Concurrent use
of fosamprenavir should be avoided owing to altered exposure to lopinavir with
decreased levels of amprenavir. Also, con-comitant use of lopinavir/ritonavir
and rifampin is contraindicated due to an increased risk for hepatotoxicity.
Since the oral solution of lopinavir/ritonavir contains alcohol, concurrent
disulfiram and metronidazole are contraindicated. There is no evidence of human
teratogenicity of lopinavir/ritonavir; short-term safety in pregnant women has
been demonstrated for mother and infant.
NELFINAVIR
Nelfinavir has high
absorption in the fed state (70–80%), under-goes metabolism by CYP3A, and is
excreted primarily in the feces. The plasma half-life in humans is 3.5–5 hours,
and the drug is more than 98% protein-bound.
The most common
adverse effects associated with nelfinavir are diarrhea and flatulence.
Diarrhea often responds to anti-diarrheal medications but can be dose-limiting.
Nelfinavir is an inhibitor of the CYP3A system, and multiple drug interactions
may occur (Tables 49–3 and 49–4). An increased dosage of nelfi-navir is
recommended when co-administered with rifabutin (with a decreased dose of
rifabutin), whereas a decrease in saquinavir dose is suggested with concurrent
nelfinavir. Co-administration with efavirenz should be avoided due to decreased
nelfinavir levels. Nelfinavir has a favorable safety and pharmacokinetic
profile for pregnant women compared with that of other PIs (Table 49–5); there
is no evidence of human teratogenicity.

RITONAVIR
Ritonavir has a high
bioavailability (about 75%) that increases with food. It is 98% protein-bound
and has a serum half-life of 3–5 hours. Metabolism to an active metabolite
occurs via the CYP3A and CYP2D6 isoforms; excretion is primarily in the feces.
Caution is advised when administering the drug to persons with impaired hepatic
function.
Potential adverse
effects of ritonavir, particularly when adminis-tered at full dosage, are
gastrointestinal disturbances, paresthesias (circumoral or peripheral),
elevated serum aminotransferase levels, altered taste, headache, and elevations
in serum creatine kinase. Nausea, vomiting, diarrhea, or abdominal pain
typically occur during the first few weeks of therapy but may diminish over
time or if the drug is taken with meals. Dose escalation over 1–2 weeks is
recommended to decrease the dose-limiting side effects. Liver adenomas and
carcinomas have been induced in male mice receiv-ing ritonavir; no similar
effects have been observed to date in humans.
Ritonavir is a potent
inhibitor of CYP3A4, resulting in many potential drug interactions (Tables 49–3
and 49–4). However, this characteristic has been used to great advantage when
ritonavir is administered in low doses (100–200 mg twice daily) in combina-tion
with any of the other PI agents, in that increased blood levels of the latter
agents permit lower or less frequent dosing (or both) with greater tolerability
as well as the potential for greater efficacy against resistant virus.
Therapeutic levels of digoxin and theophyl-line should be monitored when
co-administered with ritonavir
SAQUINAVIR
In its original
formulation as a hard gel capsule (saquinavir-H; Invirase), oral saquinavir was
poorly bioavailable (only about 4% after food). However, reformulation of
saquinavir-H for once-daily dosing in combination with low-dose ritonavir has
both improved antiviral efficacy and decreased gastrointestinal adverse
effects.
Saquinavir
should be taken within 2 hours after a fatty meal for enhanced absorption.
Saquinavir is 97% protein-bound, and serum half-life is approximately 2 hours.
Saquinavir has a large volume of distribution, but penetration into the
cerebrospinal fluid is negligible. Excretion is primarily in the feces.
Reported adverse effects include gastrointestinal discomfort (nausea, diarrhea,
abdominal discomfort, dyspepsia) and rhinitis. When administered in combination
with low-dose ritonavir, there appears to be less dyslipidemia or
gastrointestinal toxicity than with some of the other boosted PI regimens.
However, the concur-rent use of saquinavir and ritonavir is newly recognized to
confer an increased risk of QT prolongation (with torsades de pointes
arrhythmia) and PR interval prolongation.
Saquinavir
is subject to extensive first-pass metabolism by CYP3A4 and functions as a CYP3A4
inhibitor as well as a sub-strate; thus, there are many potential drug-drug
interactions (Tables 49–3 and 49–4). A decreased dose of saquinavir is
recom-mended when co-administered with nelfinavir. Increased saquina-vir levels
when co-administered with omeprazole necessitate close monitoring for
toxicities. Digoxin levels may increase if co-administered with saquinavir and
should therefore be monitored. Liver function tests should be monitored if
saquinavir is co-administered with delavirdine or rifampin. There is no
evidence of human teratogenicity from saquinavir; there is short-term safety
data for both mother and infant.
TIPRANAVIR
Tipranavir is a newer
PI indicated for use in treatment-experienced HIV-1-infected patients who
harbor strains resistant to other PI agents. It is used in combination with
ritonavir to achieve effective serum levels and is not approved for
treatment-naïve patients.
Bioavailability
is poor but is increased when taken with a high-fat meal. The drug is
metabolized by the liver microsomal system and is contraindicated in patients
with hepatic insufficiency. Tipranavir contains a sulfonamide moiety and should
not be administered to patients with known sulfa allergy.
The most common
adverse effects from tipranavir are diarrhea, nausea, vomiting, and abdominal
pain. An urticarial or maculo-papular rash is more common in women and may be
accompanied.by systemic symptoms or desquamation. Liver toxicity, including
life-threatening hepatic decompensation, has been observed and is more common
in patients with chronic HBV or HCV infection. Tipranavir should be
discontinued in patients who have increased serum transaminase levels that are
more than 10 times the upper limit of normal or more than 5 times normal in
combination with increased serum bilirubin. Because of an increased risk for
intra-cranial hemorrhage in patients receiving tipranavir/ritonavir, the drug
should be avoided in patients with head trauma or bleeding diathesis. Other
potential adverse effects include depression, eleva-tion in amylase, and
decreased white blood cell count.
Tipranavir
both inhibits and induces the CYP3A4 system. When used in combination with
ritonavir, its net effect is inhibi-tion. Tipranavir also induces
P-glycoprotein transporter and thus may alter the disposition of many other
drugs (Table 49–4). Concurrent administration of tipranavir with fosamprenavir
or saquinavir should be avoided owing to decreased blood levels of the latter
drugs. Tipranavir/ritonavir may also decrease serum levels of valproic acid and
omeprazole. Levels of lovastatin, simva-statin, atorvastatin, and rosuvastatin
may be increased, increasing the risk for rhabdomyolysis and myopathy.
Tipranavir contains a sulfonamide moiety and should be used cautiously in
patients with sulfonamide allergy.
Related Topics