Chapter: Basic & Clinical Pharmacology : Antiviral Agents
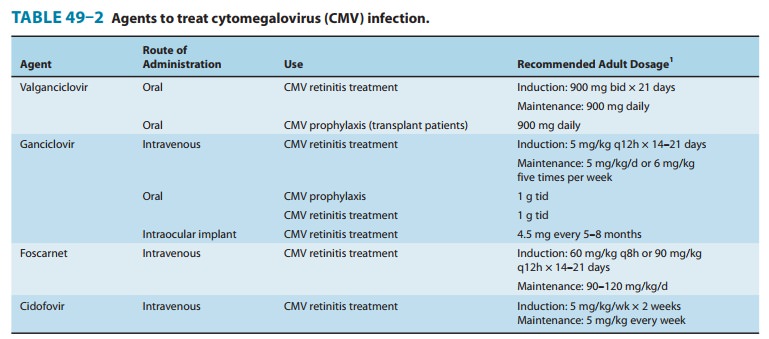
Agents to Treat Cytomegalovirus CMV Infections
AGENTS TO TREAT CYTOMEGALOVIRUS CMV INFECTIONS
CMV
infections occur primarily in the setting of advanced immu-nosuppression and
are typically due to reactivation of latent infec-tion. Dissemination of
infection results in end-organ disease, including retinitis, colitis,
esophagitis, central nervous system disease, and pneumonitis. Although the
incidence in HIV-infected patients has markedly decreased with the advent of
potent anti-retroviral therapy, clinical reactivation of CMV infection after
organ transplantation is still prevalent.
The
availability of oral valganciclovir and the ganciclovir intraoc-ular implant
has decreased the use of intravenous ganciclovir, intra-venous foscarnet, and
intravenous cidofovir for the treatment of end-organ CMV disease (Table 49–2).
Oral valganciclovir has largely replaced oral ganciclovir because of its lower
pill burden.
GANCICLOVIR
Ganciclovir
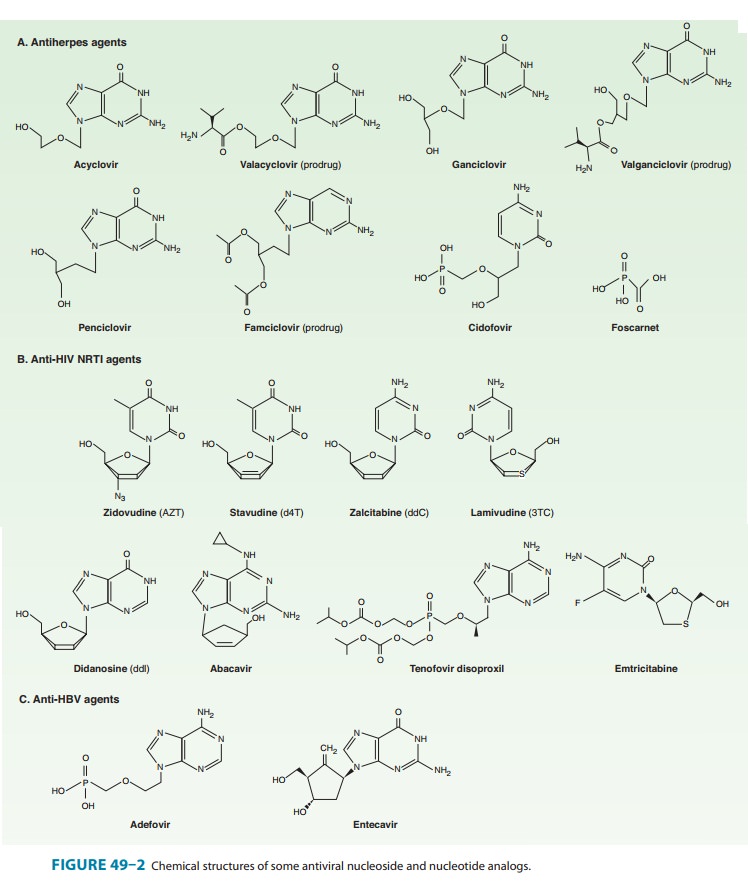
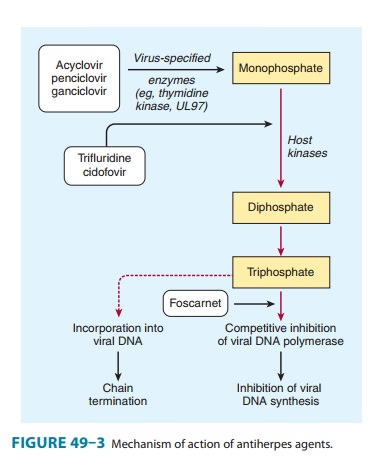
is an acyclic guanosine analog (Figure 49–2) that requires activation by
triphosphorylation before inhibiting the viral DNA polymerase. Initial
phosphorylation is catalyzed by the virus-specified protein kinase
phosphotransferase UL97 in
The activated compound competitively inhibits viral DNA polymerase and causes
termination of viral DNA elongation (Figure 49–3). Ganciclovir has in vitro
activity against CMV, HSV, VZV, EBV, HHV-6, and HHV-8. Its activity against CMV
is up to 100 times greater than that of acyclovir.
Ganciclovir
may be administered intravenously, orally, or via intraocular implant. The
bioavailability of oral ganciclovir is poor. Cerebrospinal fluid concentrations
are approximately 50% of serum concentrations. The elimination half-life is 4
hours, and the intracellular half-life is prolonged at 16–24 hours. Clearance
of the drug is linearly related to creatinine clearance. Ganciclovir is readily
cleared by hemodialysis.
Intravenous
ganciclovir has been shown to delay progression of CMV retinitis in patients
with AIDS. Dual therapy with foscarnet and ganciclovir is more effective in
delaying progression of retinitis than either drug alone (see Foscarnet),
although adverse effects are compounded. Intravenous ganciclovir is also used
to treat CMV colitis, esophagitis, and pneumonitis (the latter often treated
with a combination of ganciclovir and intravenous cytomegalovirus
immunoglobulin) in immunocompromised patients. Intravenous ganciclovir,
followed by either oral ganciclovir or high-dose oral acyclovir, reduces the
risk of CMV infection in transplant recipi-ents. Oral ganciclovir is indicated
for prevention of end-organ CMV disease in AIDS patients and as maintenance
therapy of CMV retinitis after induction. Although less effective than intrave-nous
ganciclovir, the oral form carries a diminished risk of myelo-suppression and
of catheter-related complications. The risk of Kaposi’s sarcoma is reduced in
AIDS patients receiving long-term ganciclovir, presumably because of activity
against HHV-8.
Ganciclovir may also
be administered intraocularly to treat CMV retinitis, either by direct
intravitreal injection or by intraoc-ular implant. The implant has been shown
to delay progression of retinitis to a greater degree than systemic ganciclovir
therapy. Surgical replacement of the implant is required at intervals of5–8
months. Concurrent therapy with a systemic anti-CMV agent is recommended to
prevent other sites of end-organ CMV disease.
Resistance to
ganciclovir increases with duration of usage. The more common mutation, in
UL97, results in decreased levels of the triphosphorylated (ie, active) form of
ganciclovir. The less common UL54 mutation in DNA polymerase results in higher
levels of resistance and potential cross-resistance with cidofovir and foscarnet.
Antiviral susceptibility testing is recommended in patients in whom resistance
is suspected clinically, as is the substi-tution of alternative therapies and
concomitant reduction in immunosuppressive therapies, if possible. The addition
of CMV hyperimmune globulin may also be considered.
The most common
adverse effect of systemic ganciclovir treat-ment, particularly after
intravenous administration, is myelosup-pression. Myelosuppression may be
additive in patients receiving concurrent zidovudine, azathioprine, or
mycophenolate mofetil. Other potential adverse effects are nausea, diarrhea,
fever, rash, headache, insomnia, and peripheral neuropathy. Central nervous
system toxicity (confusion, seizures, psychiatric disturbance) and
hepatotoxicity have been rarely reported. Ganciclovir is mutagenic in mammalian
cells and carcinogenic and embryotoxic at high doses in animals and causes
aspermatogenesis; the clinical signifi-cance of these preclinical data is
unclear.
Levels of ganciclovir
may rise in patients concurrently taking probenecid or trimethoprim. Concurrent
use of ganciclovir with didanosine may result in increased levels of
didanosine.
VALGANCICLOVIR
Valganciclovir
is an L-valyl ester prodrug of ganciclovir that exists as a
mixture of two diastereomers (Figure 49–2). After oral admin-istration, both
diastereomers are rapidly hydrolyzed to ganciclovir by esterases in the
intestinal wall and liver.
Valganciclovir
is well absorbed and rapidly metabolized in the intestinal wall and liver to
ganciclovir; no other metabolites have been detected. The bioavailability of
oral valganciclovir is 60%; it is recommended that the drug be taken with food. The AUC0–24h resulting from
valganciclovir (900 mg once daily) is similar to thatafter 5
mg/kg once daily of intravenous ganciclovir and approxi-mately 1.65 times that
of oral ganciclovir. The major route of elimination is renal, through
glomerular filtration and active tubular secretion. Plasma concentrations of
valganciclovir are reduced approximately 50% by hemodialysis.
Valganciclovir
is indicated for the treatment of CMV retinitis in patients with AIDS and for
the prevention of CMV disease in high-risk kidney, heart, and kidney-pancreas
transplant patients. Adverse effects, drug interactions, and resistance patterns
are the same as those associated with ganciclovir.
FOSCARNET
Foscarnet
(phosphonoformic acid) is an inorganic pyrophosphate analog (Figure 49–2) that
inhibits herpesvirus DNA polymerase, RNA polymerase, and HIV reverse
transcriptase directly without requiring activation by phosphorylation.
Foscarnet blocks the pyrophosphate binding site of these enzymes and inhibits
cleavage of pyrophosphate from deoxynucleotide triphosphates. It has in vitro
activity against HSV, VZV, CMV, EBV, HHV-6, HHV-8, HIV-1, and HIV-2.
Foscarnet is available
in an intravenous formulation only; poor oral bioavailability and
gastrointestinal intolerance preclude oral use. Cerebrospinal fluid
concentrations are 43–67% of steady-state serum concentrations. Although the
mean plasma half-life is 3–7 hours, up to 30% of foscarnet may be deposited in
bone, with a half-life of several months. The clinical repercussions of this
are unknown. Clearance of foscarnet is primarily renal and is directly
proportional to creatinine clearance. Serum drug concentrations are reduced
approximately 50% by hemodialysis.
Foscarnet
is effective in the treatment of CMV retinitis, CMV colitis, CMV esophagitis,
acyclovir-resistant HSV infection, and acyclovir-resistant VZV infection. The
dosage of foscarnet must be titrated according to the patient’s calculated
creatinine clear-ance before each infusion. Use of an infusion pump to control
the rate of infusion is important to prevent toxicity, and large volumes of
fluid are required because of the drug’s poor solubility. The combination of
ganciclovir and foscarnet is synergistic in vitro against CMV and has been
shown to be superior to either agent alone in delaying progression of
retinitis; however, toxicity is also increased when these agents are administered
concurrently. As with ganciclovir, a decrease in the incidence of Kaposi’s
sarcoma has been observed in patients who have received long-term foscarnet.
Foscarnet has been
administered intravitreally for the treat-ment of CMV retinitis in patients
with AIDS, but data regarding efficacy and safety are incomplete.
Resistance
to foscarnet in HSV and CMV isolates is due to point mutations in the DNA
polymerase gene and is typically associated with prolonged or repeated exposure
to the drug.
Mutations in the HIV-1
reverse transcriptase gene have also been described. Although
foscarnet-resistant CMV isolates are typically cross-resistant to ganciclovir,
foscarnet activity is usually main-tained against ganciclovir- and
cidofovir-resistant isolates of CMV.
Potential adverse
effects of foscarnet include renal impairment, hypo- or hypercalcemia, hypo- or
hyperphosphatemia, hypokalemia, and hypomagnesemia. Saline preloading helps
prevent nephrotox-icity, as does avoidance of concomitant administration of
drugs with nephrotoxic potential (eg, amphotericin B, pentamidine,
aminoglycosides). The risk of severe hypocalcemia, caused by chelation of
divalent cations, is increased with concomitant use of pentamidine. Genital
ulcerations associated with foscarnet therapy may be due to high levels of
ionized drug in the urine. Nausea, vomiting, anemia, elevation of liver
enzymes, and fatigue have been reported; the risk of anemia may be additive in
patients receiving concurrent zidovudine. Central nervous system toxicity includes
headache, hallucinations, and seizures; the risk of seizures may be increased
with concurrent use of imipenem. Foscarnet caused chromosomal damage in
preclinical studies.
CIDOFOVIR
Cidofovir (Figure
49–2) is a cytosine nucleotide analog with in vitro activity against CMV,
HSV-1, HSV-2, VZV, EBV, HHV-6, HHV-8, adenovirus, poxviruses, polyomaviruses,
and human papillomavirus. In contrast to ganciclovir, phosphorylation of
cidofovir to the active diphosphate is independent of viral enzymes (Figure
49–3); thus activity is maintained against thymidine kinase-deficient or
-altered strains of CMV or HSV. Cidofovir diphosphate acts both as a potent
inhibitor of and as an alternative substrate for viral DNA polymerase,
competitively inhibiting DNA synthesis and becoming incorporated into the viral
DNA chain. Cidofovir-resistant isolates tend to be cross-resistant with
ganciclovir but retain susceptibility to foscarnet.
Although the terminal
half-life of cidofovir is approximately 2.6 hours, the active metabolite cidofovir
diphosphate, has a pro-longed intracellular half-life of 17–65 hours, thus
allowing infre-quent dosing. A separate metabolite, cidofovir phosphocholine,
has a half-life of at least 87 hours and may serve as an intracellular
reservoir of active drug. Cerebrospinal fluid penetration is poor. Elimination
is by active renal tubular secretion. High-flux hemo-dialysis reduces serum
levels of cidofovir by approximately 75%.
Intravenous
cidofovir is effective for the treatment of CMV retinitis and is used experimentally
to treat adenovirus, human papillomavirus, and poxvirus infections. Intravenous
cidofovir must be administered with high-dose probenecid (2 g at 3 hours before
the infusion and 1 g at 2 and 8 hours after), which blocks active tubular
secretion and decreases nephrotoxicity. Before each infusion, cidofovir dosage
must be adjusted for alterations in the calculated creatinine clearance or for
the presence of urine protein, and aggressive adjunctive hydration is required.
Initiation of cidofovir therapy is contraindicated in patients with existing
renal insufficiency. Direct intravitreal administration of cidofovir is not
recommended because of ocular toxicity.
The primary adverse
effect of intravenous cidofovir is a dose-dependent proximal tubular nephrotoxicity,
which may be reduced with prehydration using normal saline. Proteinuria,
azotemia, metabolic acidosis, and Fanconi’s syndrome may occur. Concurrent
administration of other potentially nephrotoxic agents (eg, ampho-tericin B,
aminoglycosides, nonsteroidal anti-inflammatory drugs, pentamidine, foscarnet)
should be avoided. Prior administration of foscarnet may increase the risk of
nephrotoxicity. Other poten-tial adverse effects include uveitis, ocular
hypotony, and neutrope-nia (15–24%). Concurrent probenecid use may result in
other toxicities or drug-drug interactions . Cidofovir is mutagenic,
gonadotoxic, and embryotoxic, and caused mammary adenocarcinomas in rats.
Related Topics