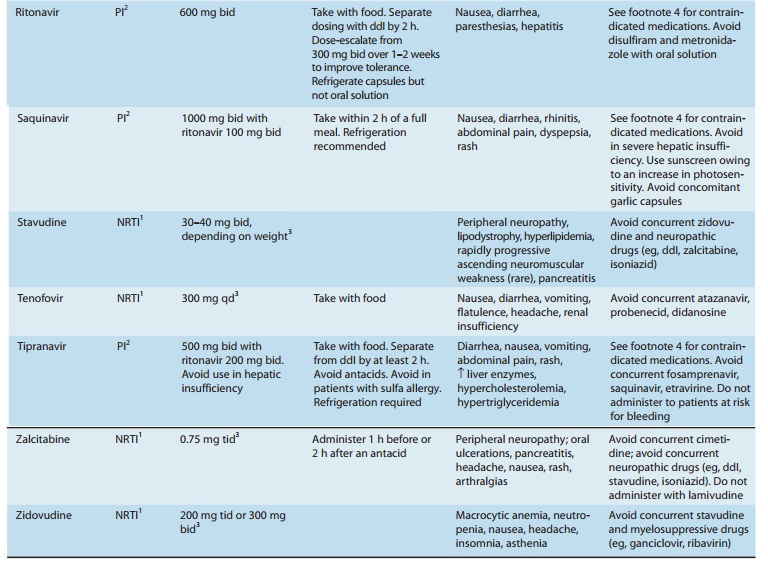
Chapter: Basic & Clinical Pharmacology : Antiviral Agents
Entry Inhibitors
ENTRY INHIBITORS
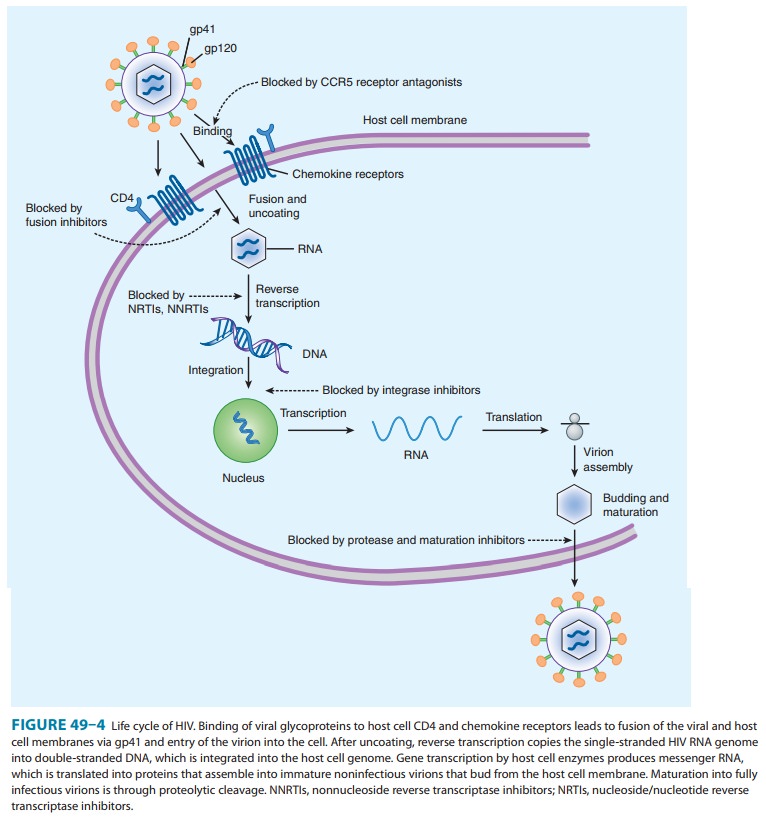
The process of HIV-1
entry into host cells is complex; each step presents a potential target for
inhibition. Viral attachment to the host cell entails binding of the viral
envelope glycoprotein com-plex gp160 (consisting of gp120 and gp41) to its
cellular receptor CD4. This binding induces conformational changes in gp120
that enable access to the chemokine receptors CCR5 or CXCR4. Chemokine receptor
binding induces further conformational changes in gp120, allowing exposure to gp41
and leading to fusion of the viral envelope with the host cell membrane and
sub-sequent entry of the viral core into the cellular cytoplasm.
ENFUVIRTIDE
Enfuvirtide
is a synthetic 36-amino-acid peptide fusion inhibitor that blocks HIV entry
into the cell (Figure 49–4). Enfuvirtide binds to the gp41 subunit of the viral
envelope glycoprotein, pre-venting the conformational changes required for the
fusion of the viral and cellular membranes. Enfuvirtide, which must be
admin-istered by subcutaneous injection, is the only parenterally adminis-tered
antiretroviral agent. Metabolism appears to be by proteolytic hydrolysis
without involvement of the CYP450 system. Elimination half-life is 3.8 hours.
Resistance
to enfuvirtide can result from mutations in gp41; the frequency and
significance of this phenomenon are being investigated. However, enfuvirtide
lacks cross-resistance with the other currently approved antiretroviral drug
classes.
The most common
adverse effects associated with enfuvirtide therapy are local injection site
reactions, consisting of painful erythematous nodules. Although frequent, these
are typically mildto moderate and rarely lead to discontinuation. Other
symptom-atic side effects may include insomnia, headache, dizziness, and
nausea. Hypersensitivity reactions may rarely occur, are of varying severity,
and may recur on rechallenge. Eosinophilia is the primary laboratory
abnormality seen with enfuvirtide administration. In prospective clinical
trials, an increased rate of bacterial pneumonia was noted in patients
receiving enfuvirtide. No drug-drug interac-tions have been identified that
would require the alteration of the dosage of concomitant antiretroviral or
other drugs.
MARAVIROC
Maraviroc
binds specifically and selectively to the host protein CCR5, one of two
chemokine receptors necessary for entrance of HIV into CD4+ cells.
Maraviroc is approved for adults with CCR5-tropic (also known as R5) HIV-1
infection who are expe-riencing virologic failure due to resistance to other
antiretroviral agents. Studies have shown that 52–60% of patients in whom at
least two antiviral regimens had failed were infected with R5 HIV. Since
maraviroc is active against HIV that uses the CCR5 co-receptor exclusively, and
not against HIV strains with CXCR4, dual, or mixed tropism, tropism testing
should be per-formed before initiating treatment with maraviroc. Clinical
experience with the use of maraviroc in treatment-naïve patients is limited.
The absorption of
maraviroc is rapid but variable, with the time to maximum absorption generally
being 1–4 hours after ingestion of the drug. Most of the drug (≥ 75%) is excreted in
the feces, whereas approximately 20% is excreted in urine. The recom-mended
dose of maraviroc varies according to renal function and the concomitant use of
CYP3A inducers or inhibitors. Maraviroc is contraindicated in patients with
severe or end-stage renal impairment who are taking concurrent CYP3A inhibitors
or inducers, and caution is advised when used in patients with preex-isting
hepatic impairment and in those co-infected with HBV or HCV. Maraviroc has been
shown to have excellent penetration into the cervicovaginal fluid, with levels
almost four times higher than the corresponding concentrations in blood plasma.
Resistance
to maraviroc is associated with one or more mutations in the V3 loop of gp120.
There appears to be no cross-resistance with drugs from any other class,
including the fusion inhibitor enfu-virtide. However, virologic failure of
regimens containing maraviroc may potentially be caused by emergence of
non–CCR5-tropic virus (eg, CXCR4-tropic virus) or by changes in viral tropism,
owing to the development of multiple mutations throughout gp160.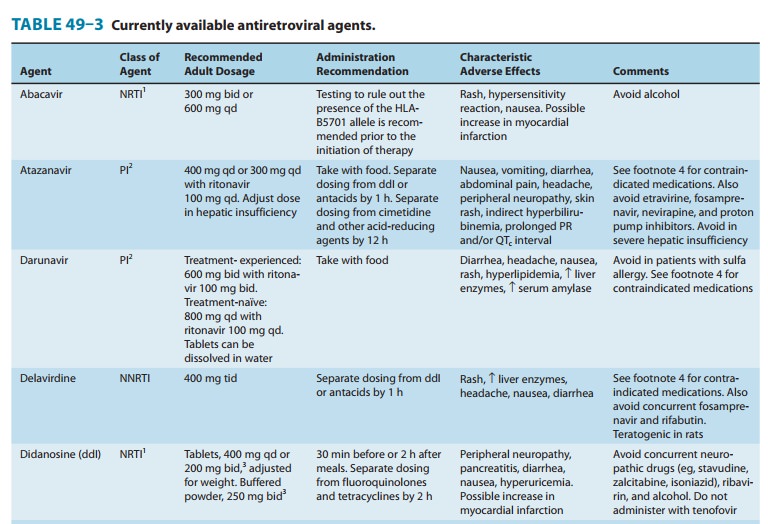
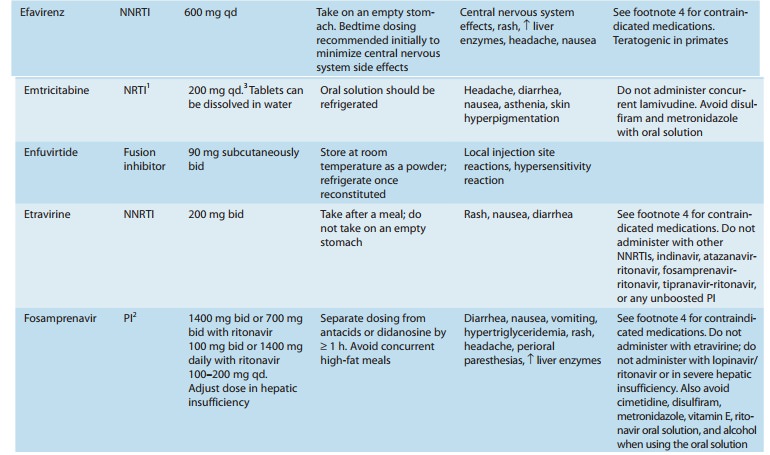
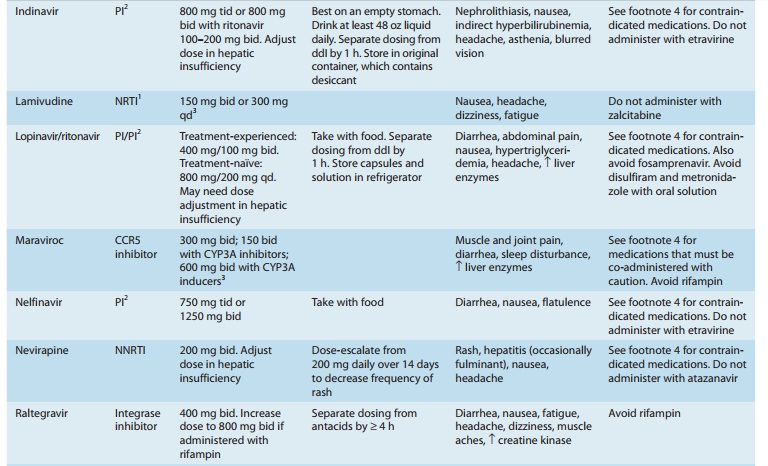
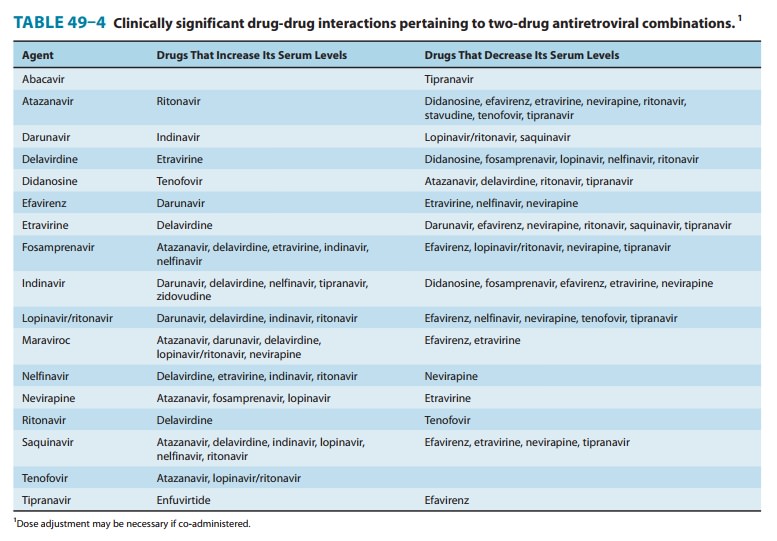
Maraviroc
is a substrate for CYP3A4 and therefore requires adjustment in the presence of
drugs that interact with these enzymes (Tables 49–3 and 49–4). It is also a substrate
for P-glycoprotein, which limits intracellular concentrations of the drug. The
dosage of maraviroc must be decreased if it is co-administered with strong
CYP3A inhibitors (eg, delavirdine, ketoconazole, itraconazole, clarithromycin,
or any protease inhibi-tor other than tipranavir) and must be increased if
co-administered with CYP3A inducers (eg, efavirenz, etravirine, rifampin,
car-bamazepine, phenytoin, or St. John’s wort).
Potential adverse
effects include cough, upper respiratory tract infections, postural hypotension
(particularly in the setting of renal insufficiency), muscle and joint pain,
abdominal pain, diarrhea, and sleep disturbance. Due to reports of
hepatotoxicity, which may be preceded by evidence of a systemic allergic
reaction (ie, pruritic rash, eosinophilia, or elevated IgE), discontinuation of
maraviroc should be considered promptly if this constellation of signs occurs.
Myocardial ischemia and infarction have been observed in patients receiving
maraviroc; therefore caution is advised in patients at increased cardiovascular
risk.
There
has been some concern that blockade of the chemokine CCR5 receptor—a human
protein—may result in decreased immune surveillance, with a subsequent
increased risk of malig-nancy (eg, lymphoma) or infection. To date, however,
there has been no evidence of an increased risk of either malignancy or
infection in patients receiving maraviroc.
Related Topics