Chapter: Plant Biochemistry: Photosynthesis is an electron transport process
The basic structure of a photosynthetic reaction center has been resolved by X-ray structure analysis
The basic structure of a photosynthetic reaction center has been resolved by X-ray structure analysis
The reaction centers of purple bacteria proved to be especially suitable objects for explaining the structure and function of the photosynthetic machinery. It was a great step forward when in 1970 Roderick Clayton (USA) developed a method for isolating reaction centers from purple bacte-ria. Analysis of the components of the reaction centers of the different purple bacteria (shown in Table 3.1 for the reaction center of Rhodobacter sphaer-oides as an example) revealed that the reaction centers had the same basic structure in all the purple bacteria investigated.
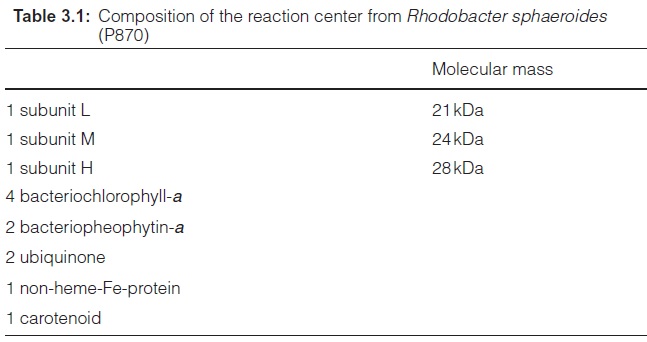
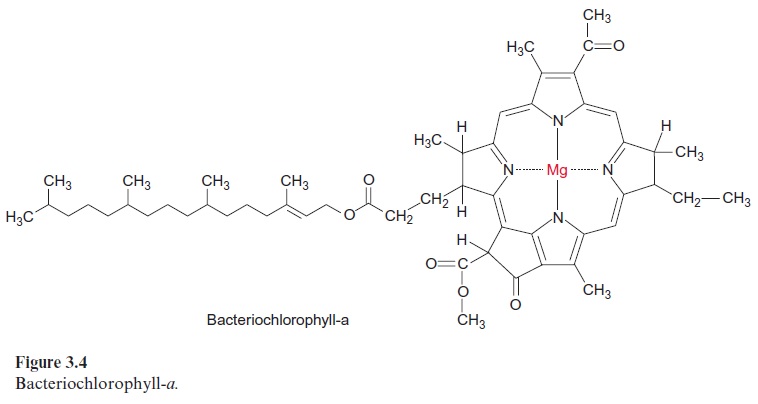
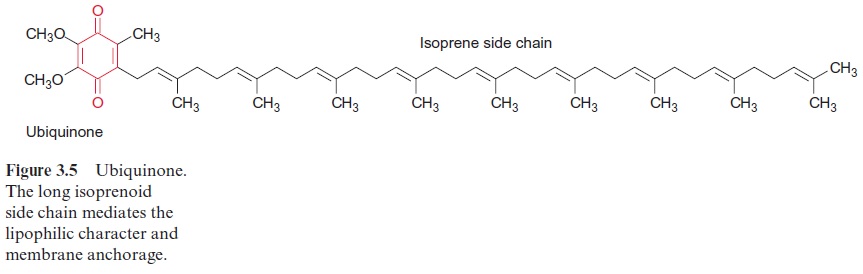
The minimum structure consists of the three subunits L, M, and H (light, medium, and heavy). Subunits L and M are peptides with a similar amino acid sequence. They are homologous. The reaction center of Rb. sphaeroides contains four bac-teriochlorophyll-a (Bchl-a, Fig. 3.4) and two bacteriopheophytin-a (Bphe-a). Pheophytins differ from chlorophylls in that they lack magnesium as the central atom. In addition, the reaction center contains an iron atom that is not part of a heme. It is therefore called a non-heme iron. Furthermore, the reaction center is comprised of two molecules of ubiquinone (Fig. 3.5), which are designated as QA and QB. QA is tightly bound to the reaction center, whereas QB is only loosely associated with it.
X-ray structure analysis of the photosynthetic reaction center
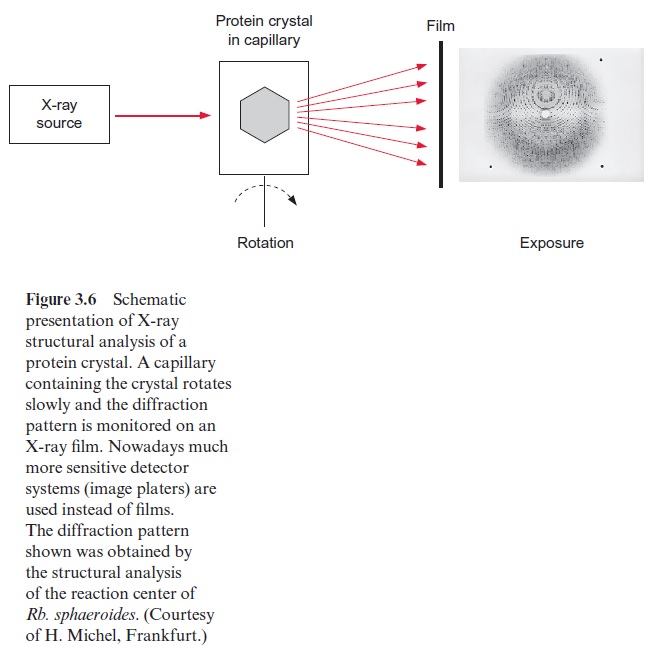
If ordered crystals can be prepared from a protein, it is possible to analyze the spherical structure of the protein molecule by X-ray structure analysis. In this method a protein crystal is exposed to X-ray irradiation. The elec-trons of the atoms in the molecule cause a scattering of X-rays. Diffraction is observed when the irradiation passes through a regular repeating struc-ture. The corresponding diffraction pattern, consisting of many single reflections, is measured by an X-ray film positioned behind the crystal or by an alternative detector. The principle is demonstrated in Figure 3.6. To obtain as many reflections as possible, the crystal, mounted in a capillary, is rotated. From a few dozen to up to several hundred exposures are required for one set of data, depending on the form of the crystal and the size of the crystal lattice. To evaluate a new protein structure, several sets of data are required in which the protein has been changed by the incorporation or binding of a heavy metal ion. With the help of elaborate computer pro-grams, it is possible to reconstruct the spherical structure of the exposed protein molecules by applying the rules for scattering X-rays by atoms of various electron densities.
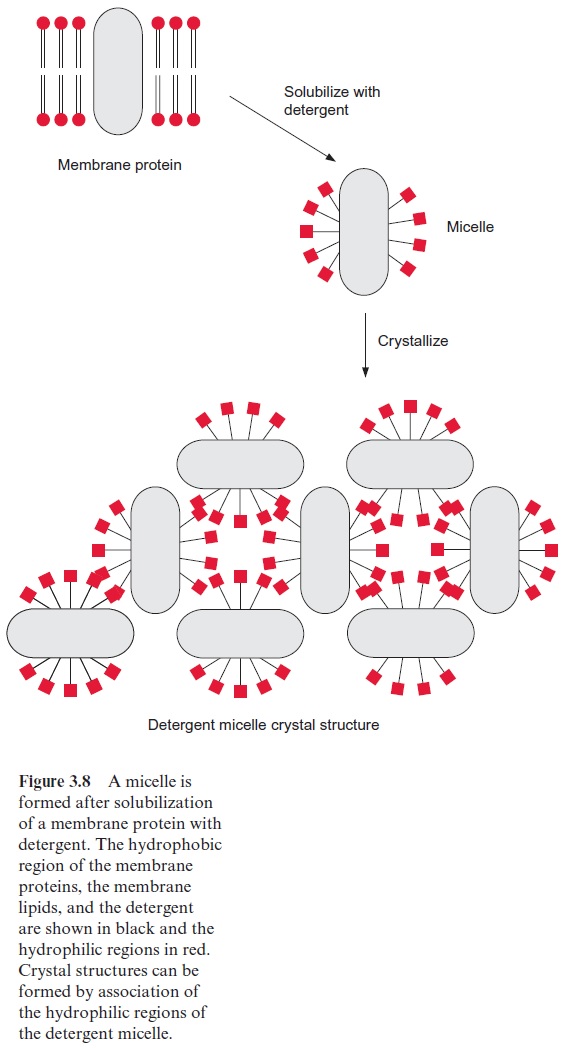
X-ray structure analysis requires a highly technical expenditure and is very time-consuming, but the actual limiting factor in the elucidation of a spherical structure is usually the preparation of suitable single crystals. Until 1980 it was thought to be impossible to prepare crystals suitable for X-ray structure analysis from hydrophobic membrane proteins. The appli-cation of the detergent N,N -dimethyldodecylamine-N-oxide (Fig. 3.7) was a great step forward in helping to solve this problem. This detergent forms water-soluble protein-detergent micelles with membrane proteins. With the addition of ammonium sulfate or polyethylene glycol is was then pos-sible to crystallize membrane proteins. The micelles form a regular lattice in these crystals (Fig. 3.8). The protein in the crystal remains in its native state since the hydrophobic regions of the membrane protein, which nor-mally border on the hydrophobic membrane, are covered by the hydropho-bic chains of the detergent.
Using this procedure, Hartmut Michel (Munich) succeeded in obtaining crystals from the reaction center of the purple bacterium Rhodopseudomonas viridis and, together with his colleagues Johann Deisenhofer and Robert Huber, performed an X-ray structure analysis of these crystals. The immense amount of time invested in these investigations is illustrated by the fact that the evaluation of the stored data sets alone took two and a half years (nowa-days modern computer programs would do it very much faster). The X-ray structure analysis of a photosynthetic reaction center successfully elucidated for the first time the three-dimensional structure of a membrane protein. For this work, Michel, Deisenhofer, and Huber were awarded the Nobel Prize in Chemistry in 1988. Using the same method, the reaction center of Rb. sphaeroides was analyzed and it turned out that the basic structures of the two reaction centers are astonishingly similar.
The reaction center of Rhodopseudomonas viridis has a symmetric structure
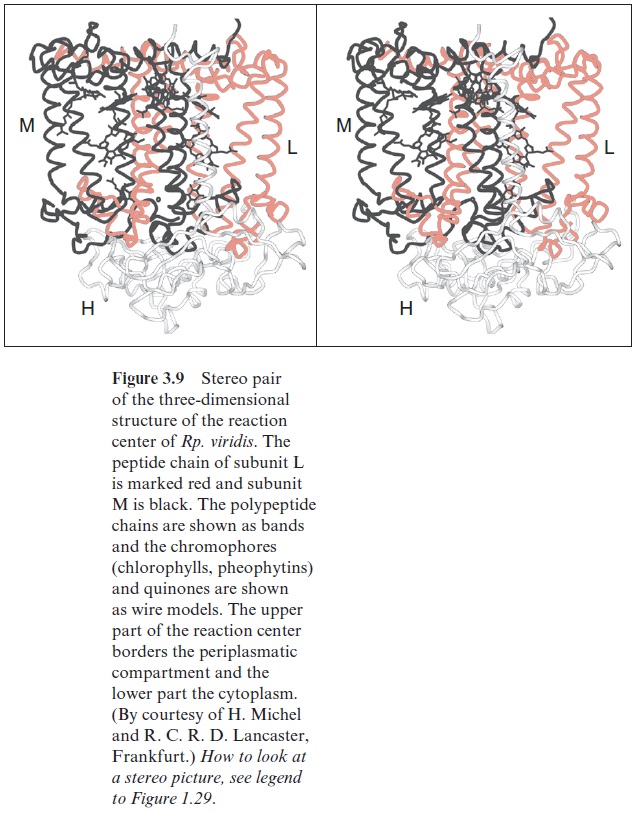
Figure 3.9 shows the three-dimensional structure of the reaction center of the purple bacterium Rhodopseudomonas viridis. The molecule has a cylin-drical shape and is about 8 nm long. The homologous subunits L (red) and M (black) are arranged symmetrically and enclose the chlorophyll and phe-ophytin molecules. The H subunit is attached like a lid to the lower part of the cylinder.
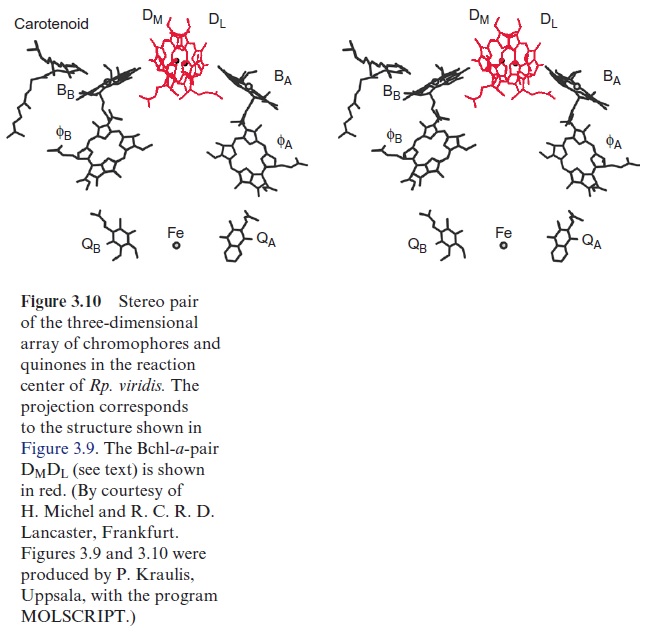
In the same projection as in Figure 3.9, Figure 3.10 shows the location of the chromophores in the protein molecule. All the chromophores are positioned as pairs divided by a symmetry axis. Two Bchl-a molecules (DM, DL) can be recognized in the upper part of the structure. The two tetrapy-rrole rings are so close (0.3 nm) that their orbitals overlap in the excited state. This confirmed the actual existence of the “special pair” of chloro-phyll molecules, postulated earlier from spectroscopic investigations, as the site of the primary redox process of photosynthesis. The chromophores are arranged underneath the chlorophyll pair in two nearly identical branches, both comprised of a Bchl-a (BA, BB) monomer and a bacteriopheophytin ( ФA, ФB). Whereas the chlorophyll pair (DM, DL) is bound by both sub-units L and M, the chlorophyll BA and the pheophytin ФA are associated with subunit L, and BB and ФB with subunit M. The quinone ring of QA is bound via hydrogen bonds and hydrophobic interaction to subunit M, whereas the loosely associated QB is bound to subunit L.
Related Topics