Chapter: Biochemistry: Nucleic Acids: How Structure Conveys Information
The Structure of DNA
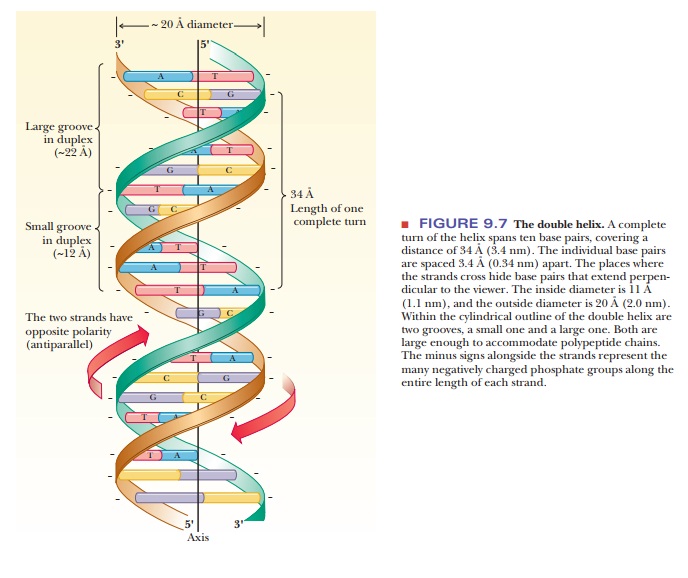
Representations
of the double-helical structure of DNA have become common in the popular press
as well as in the scientific literature. When the double helix was proposed by James Watson and Francis Crick in
1953, it touched off a flood of research activity, leading to great advances in
molecular biology.
What is the nature of the DNA double helix?
The
determination of the double-helical structure was based primarily on model
building and X-ray diffraction patterns. Information from X-ray patterns was
added to information from chemical analyses that showed that the amount of A
was always the same as the amount of T, and that the amount of G always equaled
the amount of C. Both of these lines of evidence were used to conclude that DNA
consists of two polynucleotide chains wrapped around each other to form a
helix. Hydrogen bonds between bases on opposite chains determine the alignment
of the helix, with the paired bases lying in planes perpendicular to the helix
axis. The sugar– phosphate backbone is the outer part of the helix (Figure
9.7). The chains run in antiparallel directions, one 3' to 5' and the other 5'
to 3.'
The X-ray diffraction pattern of DNA demonstrated the helical structure and the diameter. The combination of evidence from X-ray diffraction and chemi-cal analysis led to the conclusion that the base pairing is complementary, meaning that adenine pairs with thymine and that guanine pairs with cytosine. Because complementary base pairing occurs along the entire double helix, the two chains are also referred to as complementary strands.
By 1953, studies of the base composi-tion of DNA from many species had already shown that, to within experimental error,
the mole percentages of adenine and thymine (moles of these substances as
percentages of the total) were equal; the same was found to be the case with
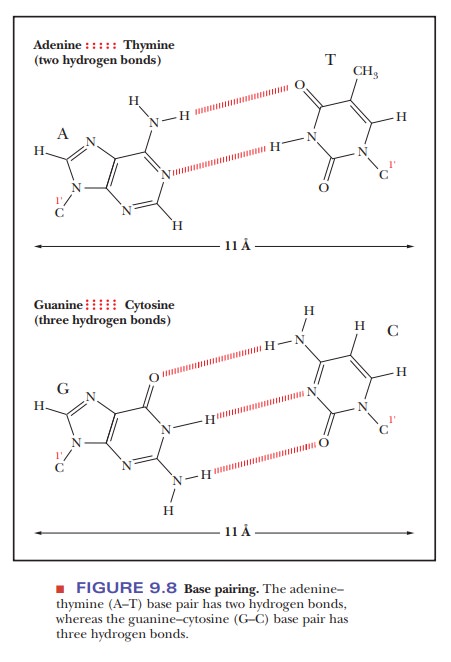
gua-nine and cytosine. An adenine–thymine (A–T) base pair has two hydrogen
bonds between the bases; a guanine–cytosine (G–C) base pair has three (Figure
9.8).
The
inside diameter of the sugar–phosphate backbone of the double helix is about 11
Ă… (1.1 nm). The distance between the points of attachment of the bases to the
two strands of the sugar–phosphate backbone is the same for the two base pairs
(A–T and G–C), about 11 Å (1.1 nm), which allows for a double helix with a
smooth backbone and no overt bulges. Base pairs other than A–T and G–C are
possible, but they do not have the correct hydrogen bonding pattern (A–C or G–T
pairs) or the right dimensions (purine–purine or pyrimidine–pyrimidine pairs)
to allow for a smooth double helix (Figure 9.8). The outside diameter of the
helix is 20 Ă… (2 nm). The length of one complete turn of the helix along its
axis is 34 Ă… (3.4 nm) and contains 10 base pairs. The atoms that make up the
two polynucleotide chains of the double helix do not completely fill an
imaginary cylinder around the double helix; they leave empty spaces known as
grooves. There is a large majorgroove and
a smaller minor groove in the double
helix; both can be sites at whichdrugs or polypeptides bind to DNA (see Figure
9.7). At neutral, physiological pH, each phosphate group of the backbone
carries a negative charge. Positively charged ions, such as Na+ or Mg2+, and
polypeptides with positively charged side chains must be associated with DNA in
order to neutralize the negative charges. Eukaryotic DNA, for example, is
complexed with histones, which are positively charged proteins, in the cell
nucleus.
Are there other possible conformations of the double helix?
The form of DNA that we have been discussing so far is called B-DNA. It is thought to be the principal form that occurs in nature.
However, other secondary structures can occur, depending on conditions such as the nature of the positive ion associated with the DNA and the specific sequence of bases. One of those other forms is A-DNA, which has 11 base pairs for each turn of the helix. Its base pairs are not perpendicular to the helix axis but lie at an angle of about 20° to the perpendicular (Figure 9.9). An important shared feature of A-DNA and B-DNA is that both are right-handed helices; that is, the helix winds upward in the direction in which the fingers of the right hand curl when the thumb is pointing upward (Figure 9.10).
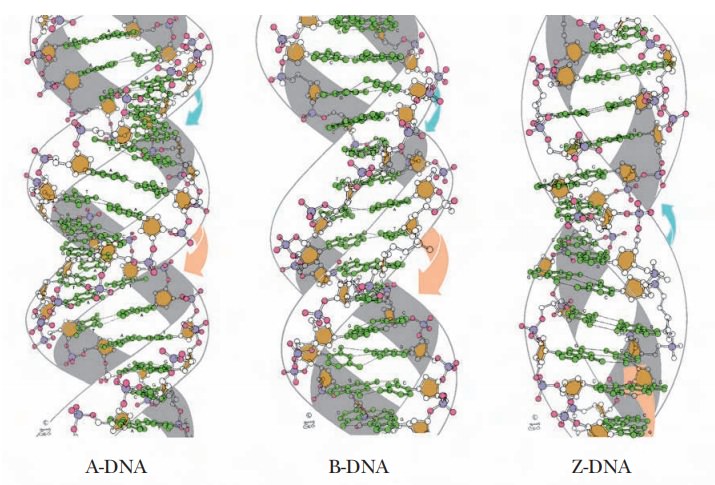
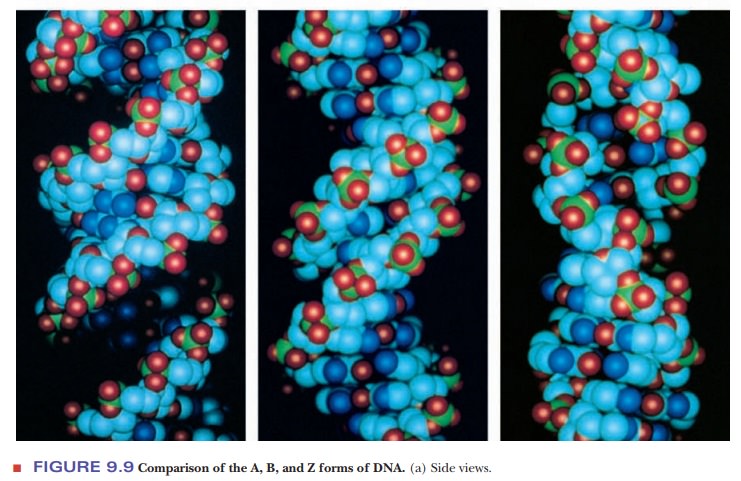
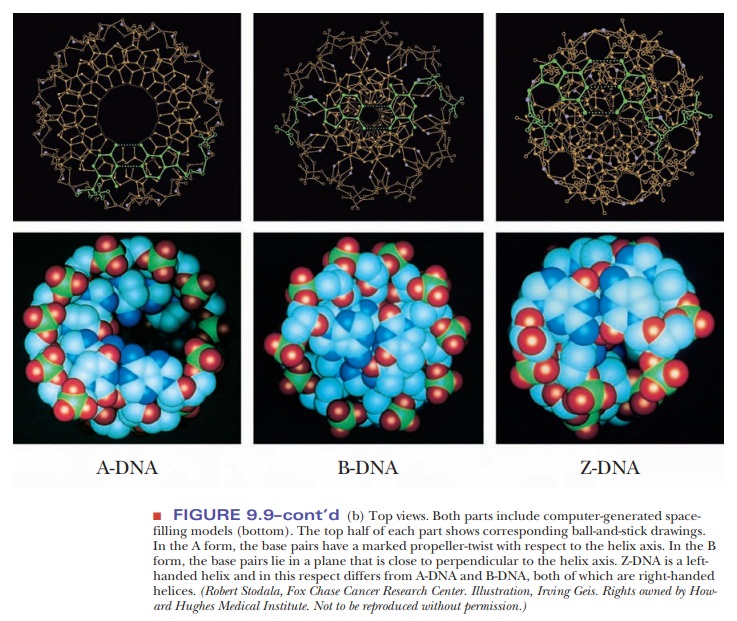
The A form of DNA was originally found in
dehydrated DNA samples, and many researchers believed that the A form was an
artifact of DNA preparation. DNA:RNA hybrids can adopt an A formation because
the 2'-hydroxyl on the ribose prevents an RNA helix from adopting the B form;
RNA:RNA hybrids may also be found in the A form.

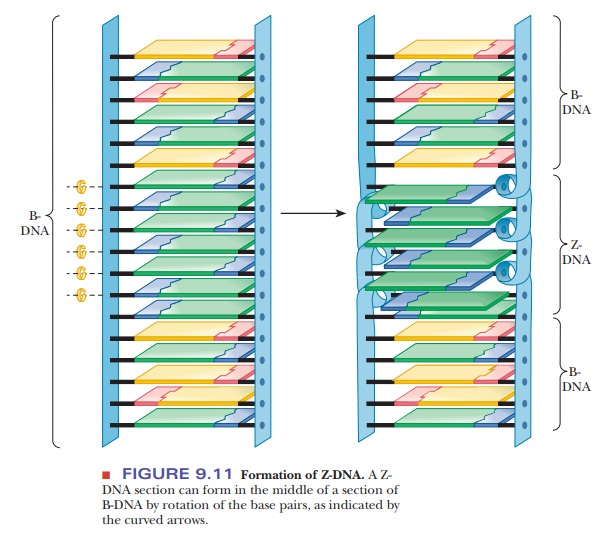
Another
variant form of the double helix, Z-DNA,
is left-handed; it winds in the direction of the fingers of the left hand
(Figure 9.10). Z-DNA is known to occur in nature, most often when there is a
sequence of alternating purine– pyrimidine, such as dCpGpCpGpCpG. Sequences
with cytosine methylated at the number 5 position of the pyrimidine ring can
also be found in the Z form. It may play a role in the regulation of gene
expression. The Z form of DNA is also a subject of active research among
biochemists. The Z form of DNA can be considered a derivative of the B form of
DNA, produced by flipping one side of the backbone 180° without having to break
either the backbone or the hydrogen bonding of the complementary bases. Figure
9.11 shows how this might occur. The Z form of DNA gets its name from the
zigzag look of the phosphodiester backbone when viewed from the side.
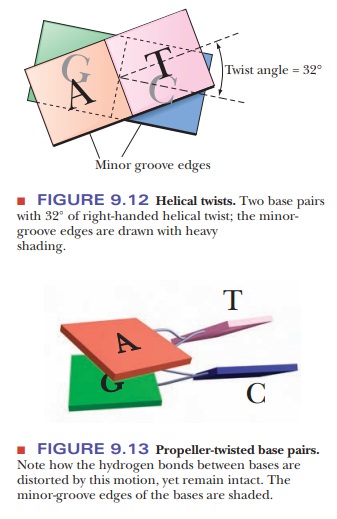
The B form of DNA has long been considered the normal, physiological DNA form. It was predicted from the nature of the hydrogen bonds between purines and pyrimidines and later found experimentally. Although it is easy to focus completely on the base pairing and the order of bases in DNA, other features of DNA structure are just as important. The ring portions of the DNA bases are very hydrophobic and interact with each other via hydrophobic bonding of their pi-cloud electrons.
This process is usually referred to as base stack-ing, and even
single-stranded DNA tends to form structures in which the basescan stack. In
standard B-DNA, each base pair is rotated 32° with respect to the preceding one
(Figure 9.12). This form is perfect for maximal base pairing, but it is not
optimal for maximal overlap of the bases. In addition, the edges of the bases
that are exposed to the minor groove must come in contact with water in this
form. Many of the bases twist in a characteristic way, called propeller-twist (Figure 9.13). In this
form, the base-pairing distances are less optimal, but the base stacking is
more optimal, and water is eliminated from the minor-groove contacts with the
bases. Besides twisting, bases also slide sideways, allowing them to interact
better with the bases above and below them. The twist and slide depends on
which bases are present, and researchers have identified that a basic unit for
studying DNA structure is actually a dinucleotide with its com-plementary
pairs. This is called a step in the
nomenclature of DNA structure. For example, in Figure 9.13, we see an AG/CT
step, which tends to adopt a dif-ferent structure from a GC/GC step. As more
and more is learned about DNA structure, it is evident that the standard B-DNA
structure, while a good model, does not really describe local regions of DNA
very well. Many DNA-binding proteins recognize the overall structure of a
sequence of DNA, which depends upon the sequence but is not the DNA sequence
itself.
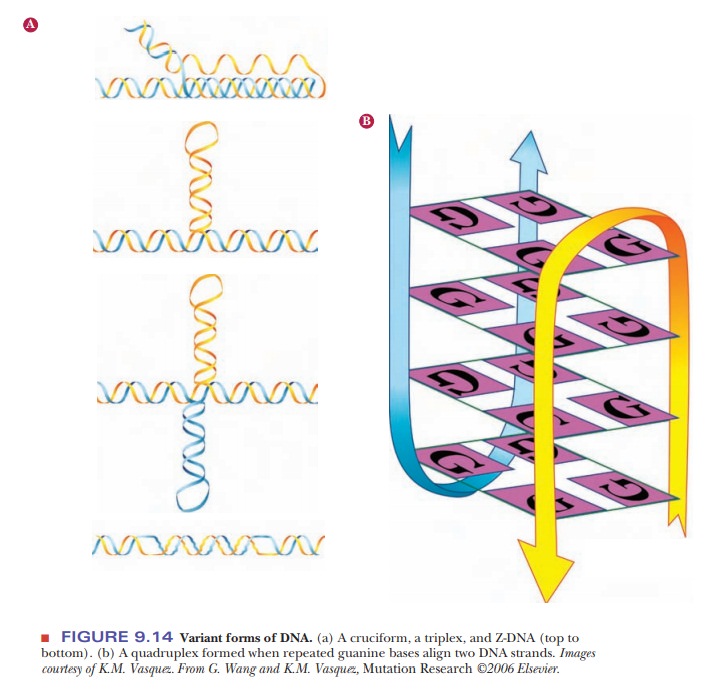
Recent
research has implicated cruciform DNA structures, triple helices, and Z-DNA in
life processes (Figure 9.14a). Suggestions have been made that quadruplex DNA
exists and that it plays a role in halting transcription (Figure 9.14b).
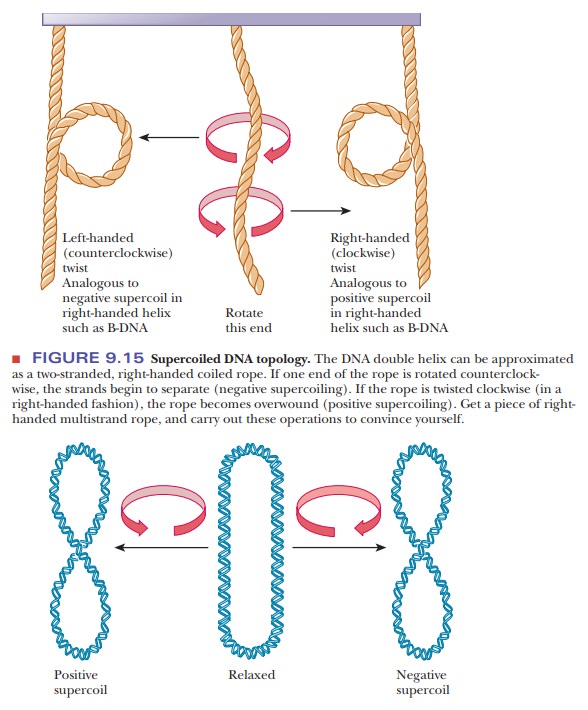
The DNA molecule has a length considerably greater than its diameter; it is not completely stiff and can fold back on itself in a manner similar to that of proteins as they fold into their tertiary structures. The double helix we have discussed so far is relaxed, which means that it has no twists in it, other than the helical twists themselves. Further twisting and coiling, or supercoiling, of the double helix is possible.
How does prokaryotic DNA supercoil into its tertiary structure?
The
first example of supercoiling we shall consider is the case of prokaryotic DNA.
If the sugar–phosphate backbone of a prokaryotic DNA forms a covalently bonded
circle, the structure is still relaxed. Some extra twists are added if the DNA
is unwound slightly before the ends are joined to form the circle. A strain is
introduced in the molecular structure, and the DNA assumes a new conformation
to compensate for the unwinding. If, because of unwinding, a right-handed
double helix acquires an extra left-handed helical twist (a supercoil), the
circular DNA is said to be negatively
supercoiled (Figure 9.15). Under different conditions, it is possible to form a
right-handed, or positively
supercoiled, structure in which there is overwinding of the closed-circle
double helix. The difference between the positively and negatively supercoiled
forms lies in their right- and left-handed natures, which, in turn, depend on
the overwinding or underwinding of the double helix.
Enzymes
that affect the supercoiling of DNA have been isolated from a vari-ety of
organisms. Naturally occurring circular DNA is negatively supercoiled except
during replication, when it becomes positively supercoiled. Cellular regulation
of this process is critical. Enzymes that are involved in changing the
supercoiled state of DNA are called topoisomerases,
and they fall into two classes. Class I topoisomerases cut the phosphodiester
backbone of one strand of DNA, pass the other end through, and then reseal the
backbone. Class II topoisomerases cut both strands of DNA, pass some of the
remaining DNA helix between the cut ends, and then reseal. In either case,
supercoils can beadded or removed. As we shall see in upcoming, these enzymes
play an important role in replication and transcription, where separation of
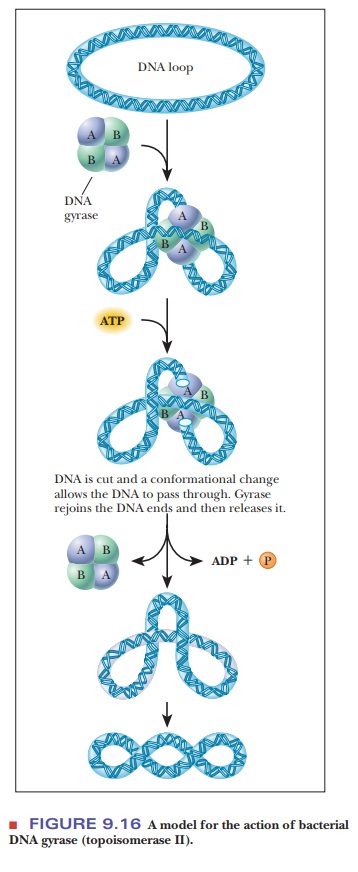
the helix strands causes supercoiling. DNA
gyrase is a bacterial topoisomerase that introduces negative supercoils
into DNA. The mechanism is shown in Figure 9.16. The enzyme is a tetramer. It
cuts both strands of DNA, so it is a class II topoisomerase.
Supercoiling
has been observed experimentally in naturally occurring DNA. Particularly
strong evidence has come from electron micrographs that clearly show coiled
structures in circular DNA from a number of different sources, including
bacteria, viruses, mitochondria, and chloroplasts. Ultracentrifugation can be
used to detect supercoiled DNA because it sediments more rapidly than the
relaxed form.
Scientists
have known for some time that prokaryotic DNA is normally circular, but
supercoiling is a relatively recent subject of research. Computer modeling has
helped scientists visualize many aspects of the twisting and knotting of
supercoiled DNA by obtaining “stop-action” images of very fast changes.
How does supercoiling take place in eukaryotic DNA?
The
supercoiling of the nuclear DNA of eukaryotes (such as plants and animals) is
more complicated than the supercoiling of the circular DNA from prokaryotes.
Eukaryotic DNA is complexed with a number of proteins, especially with basic
proteins that have abundant positively charged side chains at physiological
(neutral) pH. Electrostatic attraction between the negatively charged phosphate
groups on the DNA and the positively charged groups on the proteins favors the
formation of complexes of this sort. The resulting material is
called chromatin. Thus, topological
changes induced by supercoiling must be accommodated by the histone-protein
component of chromatin.
The principal proteins in chromatin are the histones, of which there are five main types, called H1, H2A, H2B, H3, and H4. All these proteins contain large numbers of basic amino acid residues, such as lysine and arginine. In the chromatin structure, the DNA is tightly bound to all the types of histone except H1.
The
H1 protein is comparatively easy to remove from chromatin, but dis-sociating
the other histones from the complex is more difficult. Proteins other than
histones are also complexed with the DNA of eukaryotes, but they are neither as
abundant nor as well studied as histones.
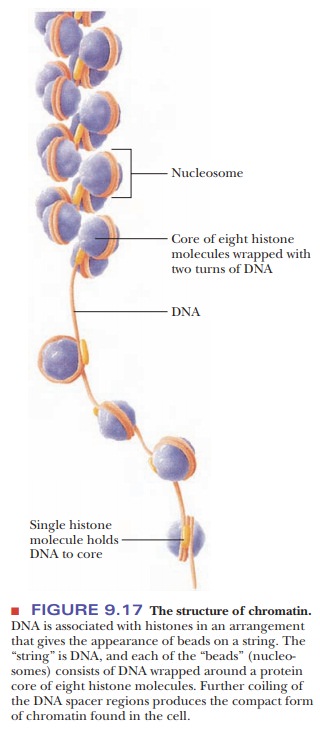
In electron micrographs, chromatin resembles beads on a string (Figure 9.17). This appearance reflects the molecular composition of the protein– DNA complex. Each “bead” is a nucleosome, consisting of DNA wrapped around a histone core. This protein core is an octamer, which includes two molecules of each type of histone but H1; the composition of the octamer is (H2A)2(H2B)2(H3)2(H4)2. The “string” portions are called spacer regions; they consist of DNA complexed to some H1 histone and nonhistone proteins. As the DNA coils around the histones in the nucleosome, about 150 base pairs are in contact with the proteins; the spacer region is about 30 to 50 base pairs long. Histones can be modified by acetylation, methylation, phosphorylation, and ubiquitinylation. Ubiquitin is a protein involved in the degradation of other proteins. Modifying histones changes their DNA and protein-binding characteristics, and how these changes affect transcription and replication is a subject of active research.
Summery
The double helix is the predominant secondary
structure of DNA. The sugar–phosphate backbones, which run in antiparallel
directions on the two strands, lie on the outside of the helix. Pairs of bases,
one on each strand, are held in alignment by hydrogen bonds.
The base pairs lie in a plane perpendicular to the helix axis in
the most usual form of the double helix, but there are variations in structure.
The
tertiary structure of DNA depends on supercoiling. In prokaryotes, the circular
DNA is twisted before the circle is sealed, giving rise to super-coiling. In
eukaryotes, the supercoiled DNA is complexed with proteins known as histones.
Related Topics