Chapter: Biotechnology Applying the Genetic Revolution: DNA Synthesis in Vivo and in Vitro
Replication of DNA
REPLICATION OF DNA
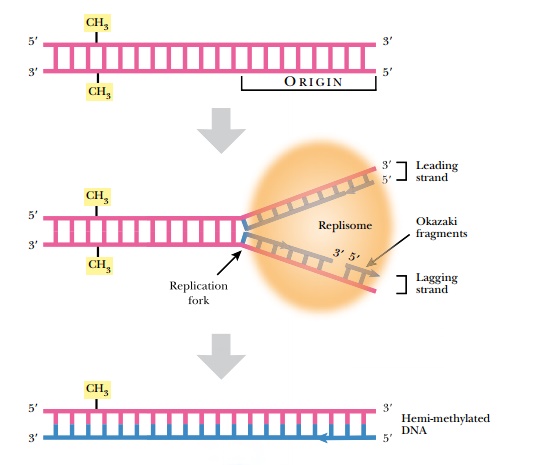
In order to maintain the integrity of an organism, the entire genome must be replicated identically. Even for gene creatures such as plasmids, viruses, or transposons, replication is critical for their survival. The complementary two-stranded structure of DNA is the key to understanding how it is duplicated during cell division. The double-stranded helix unwinds, and the hydrogen bonds holding the bases together melt apart to form two single strands. This Y-shaped region of DNA is the replication fork (Fig. 4.1). Replication starts at a specific site called an origin of replication (ori) on the chromosome. The origin is called oriC on the E. coli chromosome and covers about 245 base pairs of DNA. The origin has mostly AT base pairs, which require less energy to break than GC pairs.
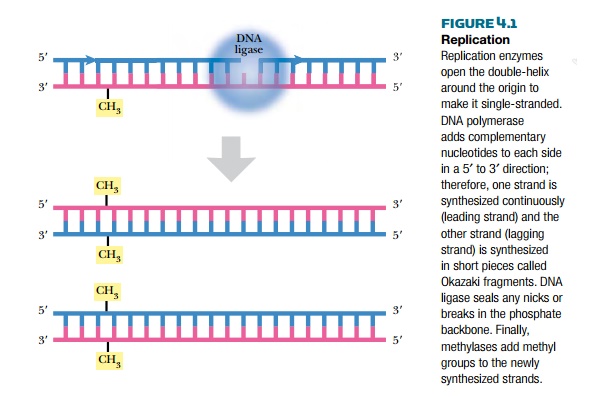
Once the replication fork is established, a large assembly of enzymes and factors assembles to synthesize the complementary strands of DNA (see Fig. 4.1). DNA polymerase starts synthesizing the complementary strand on one side of the fork by adding complementary bases in a 5′ to 3′ direction. This strand is synthesized continuously because there is always a free 3′-OH group. This strand is called the leading strand. Because DNA polymerase synthesizes only in a 5′ to 3′ direction, the other strand, called the lagging strand, is synthesized as small fragments called Okazaki fragments. The lagging strand fragments are ligated together by an enzyme called DNA ligase. Ligase links the 3′-OH and the 5′-PO4 of neighboring nucleotides, forming a phosphodiester bond. The final step is to add methyl (CH3) groups along the new strand (see later discussion). The original double-stranded helix is now two identical double-stranded helices, each containing one strand from the original molecule and one new strand. This is why the process is called semiconservative replication.
Uncoiling the DNA
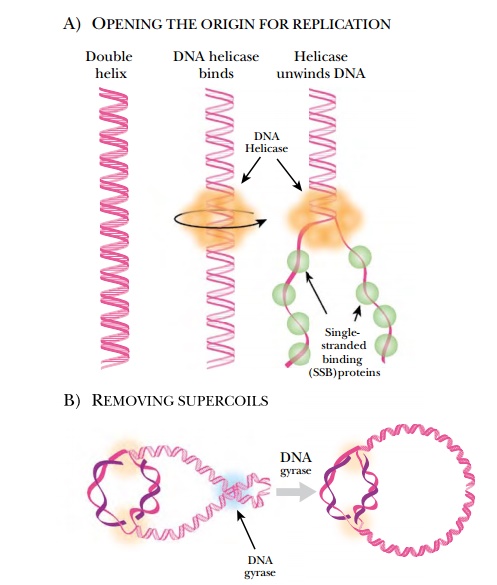
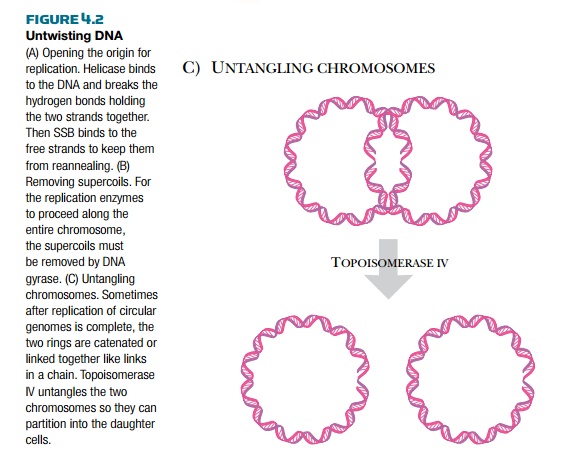
Because DNA is condensed into supercoils in order to fit inside the cell, several different enzymes are needed to open and relax the DNA before replication can start (Fig. 4.2). DNA helicase and DNA gyrase attach near the replication fork and untwist the strands of DNA. DNA gyrase removes the supercoiling, and helicase unwinds the double helix by dissolving the hydrogen bonds between the paired bases. The two strands are kept apart bysingle-stranded binding protein, which coats the single-stranded regions. This prevents the two strands from reannealing, so that other enzymes can gain access to the origin and begin replication.
As DNA polymerase travels along the DNA, more positive supercoils are added ahead of the replication fork. Because the bacterial chromosome is negatively supercoiled, initially the new positive supercoils relax the DNA. After about 5% of the genome has been replicated, though, the positive supercoils begin to accumulate and need to be removed. DNA gyrase cancels the positive supercoils by adding negative supercoils. When circular chromosomes
are replicated, the two daughter copies may become catenated, or connected like two links of a chain. Topoisomerase IV releases catenated daughter strands by introducing double-stranded nicks into one chromosome. The second copy can then pass through the first, giving two separated molecules.
Priming DNA Synthesis
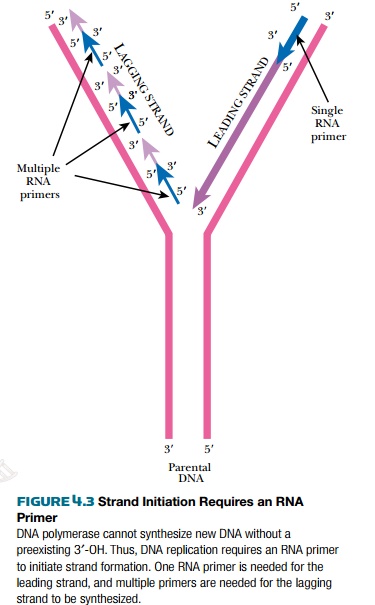
Once the origin is opened up, the replisome is assembled there. The replisome contains the enzymes for synthesizing the leading and lagging strands. The replisome also contains helicase and SSB protein, which were described earlier. Unlike RNA polymerase, DNA polymerase needs a preexisting 3′-OH to add bases to. Consequently it cannot initiate new strands of nucleic acid but can only elongate. Therefore, an 11 to 12 base-pair length of RNA (an RNA primer) is made. DNA polymerase then makes DNA starting from this RNA primer. At the origin, a protein called PriA displaces the SSB proteins so a special RNA polymerase, called primase (DnaG), can enter and synthesize short RNA primers using ribonucleotides. Primase makes a single primer at the origin of the leading strand and makes multiple primers for the lagging strand. Two molecules of DNA polymerase III bind to the primers on the leading and lagging strands and synthesize new DNA from the 3′ hydroxyls (Fig. 4.3).
Structure and Function of DNA Polymerase
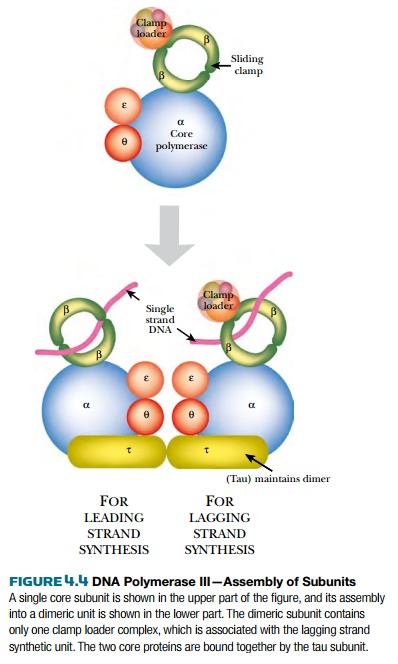
DNA polymerase III (PolIII) is the major form of DNA polymerase used to replicate bacterial chromosomes and consists of multiple protein subunits (Fig. 4.4). The sliding clamp is a donut-shaped protein consisting of a dimer of DnaN protein. Two clamps encircle the two single strands of DNA at the replication fork. A cluster of accessory proteins, the clamp loader complex, loads the clamps onto DNA strands. The two sliding clamps bind twocore enzymes, one for each strand of DNA. The core enzyme consists of three subunits, DnaE (α subunit), which links the nucleotides together, DnaQ (ε subunit), which proofreads the new strand, and HolE (θ subunit), whose role is uncertain. As the α subunit adds new nucleotides, the ε subunit recognizes any distortions and removes any mismatched bases. A correct nucleotide is then added. Bacterial DNA polymerase III can add up to 1000 bases per second, which is an extraordinarily fast rate of enzyme activity.
Synthesizing the Lagging Strand
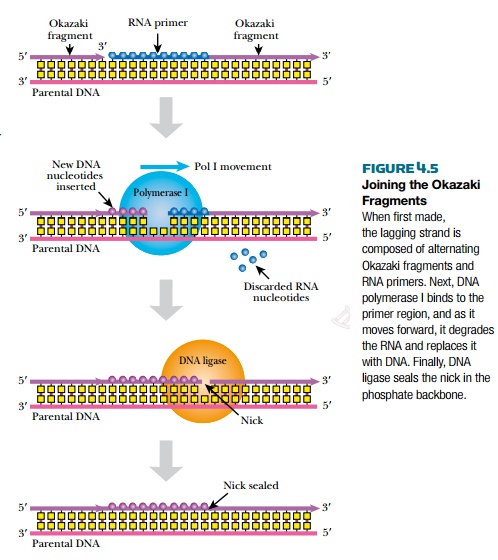
After the new lagging strand of DNA has been made, it has many segments of RNA derived from multiple RNA primers, as well as multiple breaks ornicks along the backbone that need to be sealed (Fig. 4.5). One model is that RNaseH removes the RNA primers from the lagging strand. Then DNA polymerase I fills in the regions of single-stranded DNA. Alternatively, DNA polymerase I can also excise the RNA primers itself by identifying the nicks in the DNA backbone. It then removes and replaces about 10 nucleotides downstream of each nick. Finally, the DNA fragments of the lagging strand are linked together with a ligation reaction by DNA ligase. DNA polymerase I and DNA ligase are both very important enzymes in molecular biology, and are used extensively in biotechnology.
Repairing Mistakes after Replication
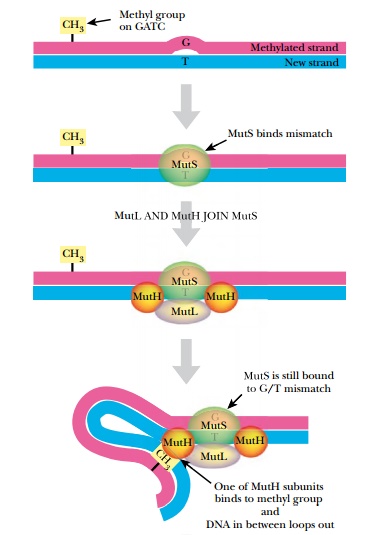
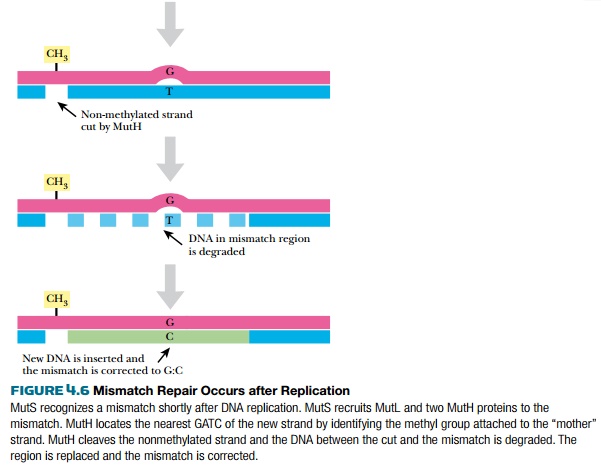
After replication is complete, the mismatch repair system corrects mistakes made by DNA polymerase. If the wrong base is inserted and DNA polymerase does not correct the error itself, there will be a small bulge in the helix at that location. Identifying which of the two bases is correct is critical. The cell assumes that the base on the new strand is wrong and the original parental base is correct. The mismatch repair system of E. coli(MutSHL) deciphers which strand is the original by monitoring methylation. Immediately after replication, the DNA is hemimethylated, that is, the old strand still has methyl groups attached to various bases but the new strand has not been methylated yet (see Fig. 4.1). Some methyl groups protect against restriction enzymes produced by the bacteria; others mark the parental strand of DNA. Two different E. coli enzymes add the latter type of methyl groups: DNA adenine methylase (Dam) adds a methyl group to the adenine in GATC, and DNA cytosine methylase (Dcm) adds a methyl group to the cytosine in CCAGG or CCTGG. These enzymes methylate the new strand after replication, but they are slow. This allows mismatch repair to find and fix any mistakes first.
Three genes of E. coli are responsible for mismatch repair, mutS,mutL, and mutH (Fig. 4.6). MutS protein recognizes the bulge or distortion in the sequence. MutH finds the nearest GATC site and nicks the nonmethylated strand—that is, the newly made strand. MutL holds
Related Topics