Chapter: Biotechnology Applying the Genetic Revolution: DNA Synthesis in Vivo and in Vitro
In Vitro Synthesis of DNA Can Determine the Sequence of Bases
IN VITRO SYNTHESIS OF DNA CAN
DETERMINE THE SEQUENCE OF BASES
Being able to quickly and easily determine the sequence of any gene has been the driving force for the recent advances made in biotechnology. Before sequencing became commonplace, identifying the gene responsible for a particular trait or disease was challenging. Frederick Sanger developed a method for sequencing a gene in vitro in 1974. He was interested in the amino acid sequence of insulin and decided to deduce the sequence of the protein from the nucleotide sequence. He invented the chain termination sequencing method, whose basic principles are still used today (Fig. 4.17). Much like DNA replication, chain termination sequencing requires a primer, DNA polymerase, a single-stranded DNA template, and deoxynucleotides. During in vitro sequencing reactions, these components are mixed and DNA polymerase makes many copies of the original template. The trick needed to deduce the sequence is to stop synthesis of some DNA chains at each base pair. Consequently, the fragments generated will differ in size by one base pair. These are then separated by gel electrophoresis. If the final base pair for each fragment is known, the sequence may be directly read from the gel (reading from bottom to top).
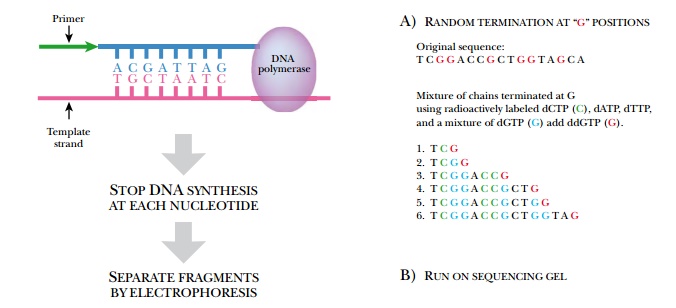
But how do we know what the
final base is for each fragment on the sequencing ladder?
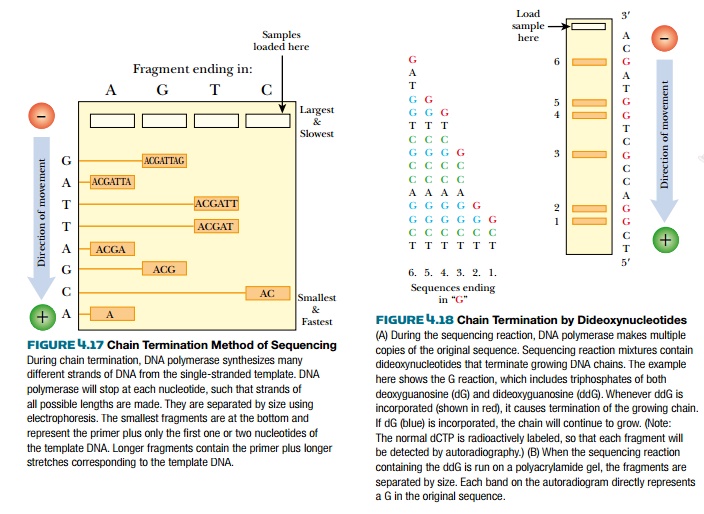
DNA polymerase synthesizes a new strand of DNA based on the template sequence. The chain consists of deoxynucleotides, each with a hydroxyl group at the 3′ position on the deoxyribose ring. DNA polymerase adds the next nucleotide by linking its 5′-phosphate to the 3′-hydroxyl of the previous nucleotide. If a nucleotide lacks the 3′-hydroxyl, no further nucleotides can be added and the chain is terminated (Fig. 4.18). During a sequencing reaction, a certain percentage of nucleotides with no 3′-hydroxyl, called dideoxynucleotides, are mixed with the normal deoxynucleotides. Historically, the dideoxynucleotides were labeled with 32P-phosphate, which is thus incorporated into each strand at the end. To get a stronger signal, one of the four normal deoxynucleotides is now radioactively labeled with 32P-phosphate. The radioactive label on the fragments is detected by autoradiography of the sequencing gel. To identify the end nucleotide, four separate reactions are run for each template, each reaction getting one of the four possible dideoxynucleotides. For example, reaction 1 contains all the normal deoxynucleotides plus a fraction of dideoxyguanine; therefore, all fragments synthesized in this mixture will end at a guanine in the sequence. Reaction 2 is similar, but contains dideoxycytosine as the terminating nucleotide and hence stops the chains at each of the cytosines. Each reaction has identical template DNA and identical primer so that each segment starts at the same location. Only the stopping points are different. Such reactions typically have a maximum length of about 300 nucleotides.
The fragments are relatively
small for DNA and vary in length by only one nucleotide; therefore, they must
be separated by size using polyacrylamide gel electrophoresis. The principle is
the same as for agarose gel electrophoresis, but polyacrylamide has smaller
pores, and so smaller fragments can be separated. Each reaction is run in a
different lane, side by side to each other. The fragments are then visualized
by autoradiography. When the sequencing gel is dried and exposed to
photographic film, the dark bands represent each of the terminated fragments.
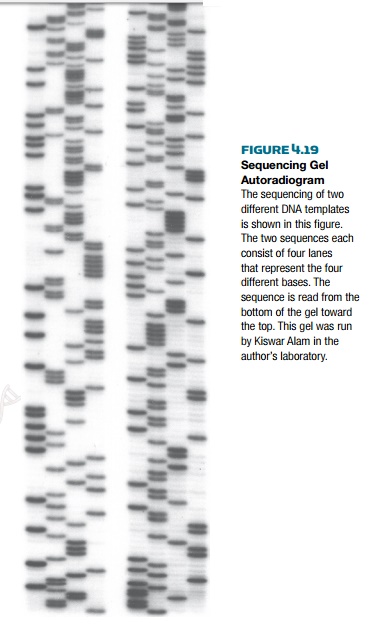
The sequence is actually read from the bottom of the gel to the top, because
the fragments terminated closest to the primer are smaller (hence run faster)
than the ones further from the primer. The bands appear as a ladder, each
separated by one nucleotide; therefore, each band is a different nucleotide on
the sequence (Fig. 4.19).
The nature of the template
DNA affects whether or not readable sequence can be obtained. Single-stranded
template DNA provides the best results and can be made by cloning the template
DNA into bacteriophage M13, which has single-stranded DNA. When the recombinant
virus infects its host,
E. coli, rolling circle replication and viral packaging create thousands
of single-stranded copies of the
template for sequencing. Alternatively, the sequence of interest is cloned into
a typical plasmid vector, and the double-stranded template is denatured either
with heat or chemicals to make it single-stranded.
Natural DNA polymerase has
been modified from its original form to enhance in vitro sequencing. Native DNA polymerase I has a domain that
excises any mismatched or defective
bases and replaces them. This activity would remove dideoxynucleotides rather
than incorporate them. This repair domain is easily removed from purified DNA
polymerase by protease digestion without affecting the other activities of DNA
polymerase. The resulting polymerase is called Klenow polymerase. Further
modifications of DNA polymerase have streamlined it for sequencing. For
example, sequencing polymerase has been engineered to increase processivity,
that is, the polymerase is less likely to fall off the template and can make
longer strands.
Related Topics