Chapter: Biotechnology Applying the Genetic Revolution: DNA Synthesis in Vivo and in Vitro
Chemical Synthesis of DNA
CHEMICAL
SYNTHESIS OF DNA
Making DNA chemically rather than biologically was one of the first new technologies to be applied by the biotechnology industry. The ability to make short synthetic stretches of DNA is crucial to using DNA replication in laboratory techniques. DNA polymerase cannot synthesize DNA without a free 3′-OH end to elongate. Therefore, to use DNA polymerase in vitro, the researcher must supply a short primer. Such primers are used to sequence DNA (see later discussion), to amplify DNA with PCR (see later discussion), and even to find genes in library screening. So a short review of how primers are synthesized is included here.

Research into chemical
synthesis of DNA began shortly after Watson and Crick published their research
on the crystal structure of DNA. H. Gobind Khorana at the University of Chicago
was an early pioneer in the study of oligonucleotide
synthesis. Technically, oligonucleotides are any piece of DNA less than 20
nucleotides in length, but today, oligonucleotide
denotes a short piece of DNA that is chemically synthesized.
In 1970, Khorana’s lab synthesized
an active tRNA molecule of 72 nucleotides (Agarwal et al., 1970). The chemistry he used was inefficient and
cumbersome, but some of his ideas are still used in current oligonucleotide
synthesis. Today chemical synthesis is done with an automated DNA synthesizer that creates DNA by
sequentially adding one nucleotide after another in the sequence specified.
Unlike in vivo DNA synthesis, artificial synthesis is done in the 3′ to 5′ direction. The first step is attaching the
first nucleotide to a porous glass bead made of controlled pore glass (CPG). The first nucleotide is not attached
directly, but linked to the bead via a spacer molecule that bonds to the 3′-OH of the nucleotide (Fig. 4.11). Many beads
are held in a column so that reagents can be washed through and removed easily.
(Using CPG is one improvement over
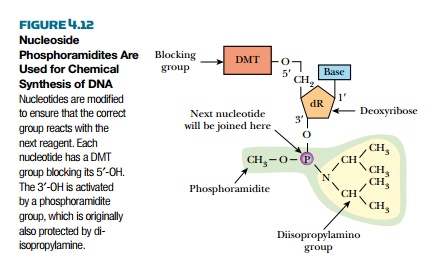
Each nucleotide is added as
its phosphoramidite, which consists
of a blocking group protecting a 3′-phosphite group (Fig. 4.12). (One problem with
early oligonucleotide synthesis technology was branching. Rather than the
incoming nucleotide adding to the 5′ end, it sometimes attached to the phosphate
linking two nucleotides.) Nowadays every added nucleotide has a
di-isopropylamine group attached to a methylated 3′ phosphite group. This structure protects
against unwanted branching. It is also stable and can be stored for long
periods. Before adding another nucleoside, the 3′ phosphite group is activated by tetrazole. The
next nucleotide is then added, and reacts with the phosphite to form a
dinucleotide (Fig. 4.13).
If the terminal nucleotide of
a growing chain fails to react with an incoming nucleotide, the chain must be
capped off to prevent generation of an incorrect sequence by later reactions.
The 5′-OH of all unreacted nucleotides is acetylated with acetic anhydride and
dimethylaminopyridine. This terminates the chain, so that no other nucleoside
phosphoramidites can be added.
Khorana’s technology. He used
polymer beads to couple the reaction, but found that the polymer swelled as the
reagents passed through the column, which inhibited synthesis. CPG is superior
because it does not swell.)
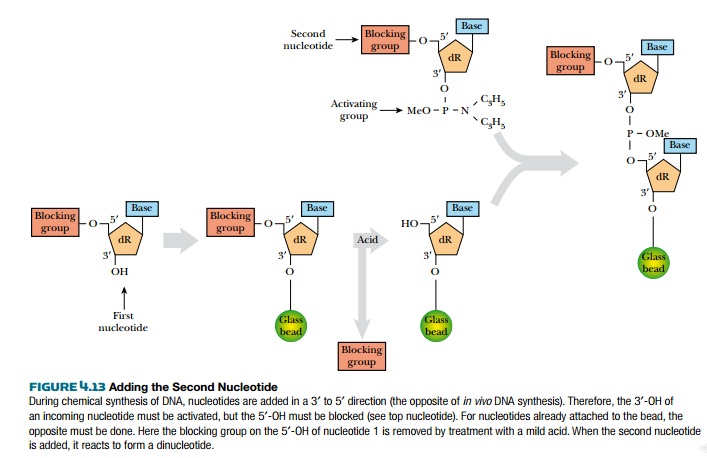
When linking the spacer to
the nucleotide 3′-OH, a chemical blocking
group is attached to the 5′-OH. Thus the 3′-OH is the only available reactive group.
Khorana’s early synthesis was revolutionary in this respect because he chose
the dimethyloxytrityl (DMT) group,
which is still used as a blocking group in today’s synthesizers. DMT has a
strong orange color and is easily removed from the 5′-OH so that another nucleotide can be linked to
the first. In practice, the CPG–spacer–first nucleotide is washed, and then the
DMT group is removed by mild acid such as trichloroacetic acid (TCA). The 5′-OH is now ready to accept the next nucleotide.
The efficiency of removing DMT is critical. If DMT is not removed completely,
many of the potential oligonucleotides will fail to elongate. The orange color
reveals the efficiency of removal and is easily measured optically.
Now the column has
CPG–spacer–first nucleoside–phosphite–second nucleoside–DMT.
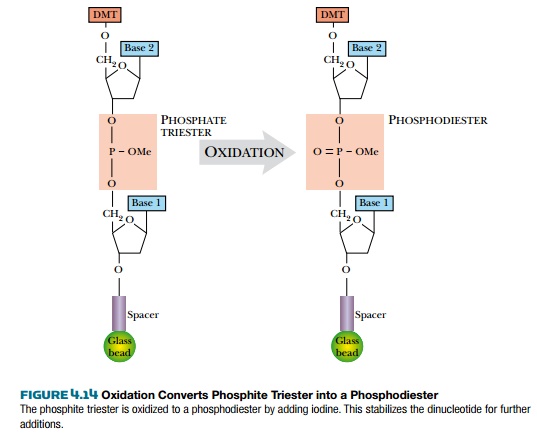
Phosphites are used because
they react much faster, but they are unstable.
Adding iodine oxidizes the
phosphite triester into the normal phosphodiester, which is more stable under
acidic conditions (Fig. 4.14).
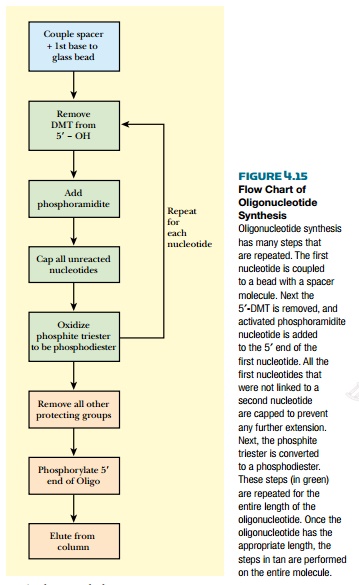
The column can now be prepared to add the third nucleotide. The DMT is removed with TCA, and the third phosphoramidite is added. The chains are capped so that any dinucleotides that didn’t react with the third phosphoramidite are prevented from adding any more nucleosides. Finally, the phosphate triester is oxidized to phosphodiester. This process continually repeats until all the desired nucleotides are added and the final oligonucleotide has the correct sequence (Fig. 4.15).
After the final
phosphoramidite nucleoside is added, the oligonucleotide still has DMT
protecting the 5′-OH, methyl groups attached
to the phosphates, and amino-protecting groups on the bases. (Amino groups
would react with the reagents during synthesis; therefore, chemical groups are
added to protect the bases before they are added to the column.) All three
types of protective groups must be removed before the oligonucleotides are
released from the column. Finally, to make the oligonucleotides biologically
active, the 5′-OH must be phosphorylated.
Usually, kinase from bacteriophage T4 is used to transfer a phosphate group
from ATP to the 5′ end of the oligonucleotides.
The newly synthesized oligonucleotide is now ready for use.
Related Topics