Chapter: Biology of Disease: Membrane, Organelle and Cytoskeletal Disorders
Plasma Membrane Disorders
PLASMA MEMBRANE DISORDERS
Numerous clinical conditions are associated with defective receptors of the plasma membrane and/or subsequent defective signal transduction or defec-tive transport of materials across the membrane. Defects of receptor proteins and signal transduction can result in the development of cancerous states , while cases of type 2 diabetes are associated with an ineffec-tive insulin receptor . Familial hypercholesterolemia, which was described, arises from defective receptors for LDL particles. The rare Tangier disease mentioned is caused by mutations in the gene for cholesterol transport protein of the plasma membrane. This protein is one of a group of transporters called ATP-binding cassette (ABC) proteins. These are multidomain structures: all have two cytosolic ATP-bind-ing domains or cassettes and two transmembrane domains each consisting of several helices. However, the most studied of the ABC transporters is the transmembrane conductance regulator protein, which regulates a chloride channel. Unlike Tangier disease, which is extremely rare, defects with this transporter lead to the much more common condition, cystic fibrosis.
CYSTIC FIBROSIS
Cystic fibrosis (CF) is a multisystem, inherited disorder characterized by the secretion of very viscous mucus secretions in the lungs, digestive tract and associated organs, and epididymis. This results in chronic respiratory disease, malabsorption, cirrhosis and electrolyte disturbances. The clinical features of CF are shown in Table 16.1.
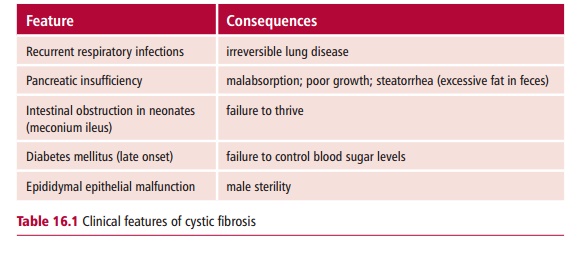
Cystic fibrosis is the commonest fatal, homozygous recessive disorder of the Caucasian population affecting about one in 2000 people in the UK, with one in 20 Caucasians carrying one copy of the mutated gene. The prevalence of CF varies throughout the world with certain populations reporting higher incidences. The onset may be at birth or later in childhood. Cystic fibrosis is characterized by decreased permeability of the apical membrane of epi-thelial tissues lining the lungs and other organs to chloride ions (Cl–) that results from mutations in the CFTR gene that encodes the cystic fibrosis transmembrane conductance regulator (Figure 16.3). This protein is a
cyclic AMP-regulated chloride channel and has been identified, cloned and sequenced and most of its mutations are known. The decreased permeability results in many exocrine secretions with altered ion and reduced water con-tents and high viscosities. The appropriate viscosity is essential for normal functions of lubrication, digestion and protection by proteins secreted at epithelial surfaces. The highly viscous secretions can cause obstructions in ducts and lead to an accumulation of mucus and colonization by bacteria. Considering that CF is essentially a fatal disease, one may wonder why the mutated genes persist in the population. One theory is that mutations inCFTR increase resistance to cholera. The toxin of Vibrio cholerae (Figure 16.4) binds to surface receptors on the plasma membranesof gut enterocytes, leading to a voluminous secretion of fluid and Cl– result-ing in a secretory diarrhea that can be fatal if untreated. Cystic fibrosis mice that do not express cftr do not secrete fluid in response to cholera toxin. Heterozygous CF mice that express half the normal amount of cftr protein secrete about half the normal fluid and Cl– in response to cholera toxin. Thus CF heterozygotes might have a selective advantage in being resistant to cholera.
Diagnosis and treatment of cystic fibrosis
A diagnosis of CF can be confirmed by demonstrating a concentration of Cl– in sweat higher than 70 mmol dm–3. Pilocarpine is first applied topically to patients to stimulate the flow of sweat, samples of which are then collected and analyzed for Cl–.
A number of screening tests for CF are now available based on the effects of CF on pancreatic function. The most popular of these relies on measurement of immunoreactive trypsin (IRT) in dried blood spots, similar to those col-lected for PKU Immunoreactive trypsin is greatly increased in infants with CF in the first month of life compared with that of healthy infants. However, the test cannot be used after the first few weeks of life since IRT levels fall as pancreatic insufficiency develops.
The identification of mutations in the CFTR of a cystic fibrosis patient will confirm the diagnosis and it can also assist with carrier testing of the relatives of the patient. It is also used to give an accurate and rapid prenatal diagnosis of CF once the parental genotypes have been confirmed. Currently there are over 1300 mutations listed in theCFTR mutation database with the largest number (500) being attributed to missense mutations. When it is not possible to detect mutant CFTR alleles, then the sweat test remains the definitive test for diagnosis of CF but, unfortunately, the sweat test is not always reliable in the first six weeks of life or in adulthood. Furthermore, some patients with CF, including a number with severe lung disease, give a normal sweat test.
The techniques for genetic analysis of CF are based on the detection of an alteration in the DNA sequence of the CTFR gene. In principle, any method capable of detecting a change in the DNA sequence can be used in mutation detection. Current techniques rely in one way or another on the hybridization of two single strands of DNA and on the ability of changes in the sequences of DNA to alter the kinetics of hybridization. These techniques depend on the polymerase chain reaction or PCR and this, in turn, requires that the normal sequence of the gene, including its intron–exon boundaries be known, as is the case for CF. Polymerase chain reaction primers are designed that will bind to adjacent regions of the gene of interest and allow it to be amplified. In most cases, whole exons are ampli-fied together with a short region of their neighboring intron sequences to give a PCR product 300 to 500 base pairs long. These products are then analyzed for specific point mutations or for generalized, nonspecific evidence of a sequence change. The testing strategy may use both approaches depending on the purpose of the analysis.
The commonest CF mutation is called ∆F508. This is the deletion of a codon, in exon 10 of the CFTR gene, that codes for a phenylalanine (F) residue at position 508 in the amino acid sequence of the encoded protein. The simplest way to detect this mutation (Figure 16.5) is by amplifying that part of the gene
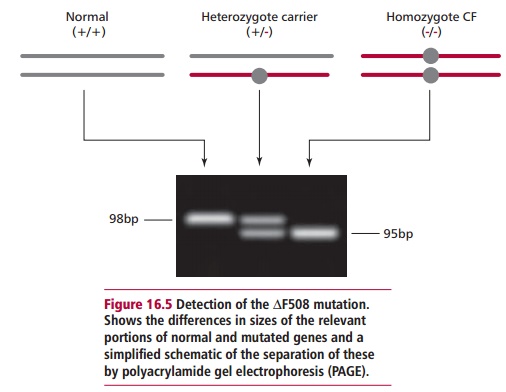
and looking for differences in the sizes of the PCR products. Polyacrylamide gel electrophoresis (PAGE) can be used to separate the products of PCR of the small fragment of exon 10 of the CFTR gene and these may be visualized with ethidium bromide or silver staining. The primers most often used are called C16B and C16D and generate a 98bp product of exon 10. However, if the ∆F508 mutation is present, the PCR product is only 95bp long. This is a relatively small difference but careful PAGE can easily separate them. This test is not absolutely specific for ∆F508 given that other 3bp deletions in this region would produce the same difference in size. Thus the mutation∆I507, the second identified CF mutation, also gives a 95bp PCR product. Mutations other than ∆F508, however, can be identified in many cases because they produce differing banding patterns following PAGE.
The treatment for cystic fibrosis is aimed at preventing respiratory infections by regular physiotherapy to try to remove the mucus, along with the use of anti-biotics to counter bacterial infections . Pancreatic enzymes, in tablet form, are given with food to counter the effects of pancreatic insufficiency.
Mice with deletions of cftr, that is gene knockout mice, have been bred to use as experimental models to test possible therapies. However, such mice do not develop lung disease in the same manner as humans and therefore do not provide an effective animal model. This has hampered experimental gene therapy studies that have tried to insert the normal gene into the lung epithelial cells of CF patients by using DNA-containing liposomes. To date, none of these therapies have been successful. Cystic fibrosis used to be fatal by the age of about 20 years but improvements in treatment mean that many sufferers now live to be 30 years and over. However, although the prognosis for CF has improved, many patients still die in early adult life.
Related Topics