Chapter: Biology of Disease: Membrane, Organelle and Cytoskeletal Disorders
Mitochondrial Disorders: Causes, Diagnosis and Treatment
MITOCHONDRIAL DISORDERS
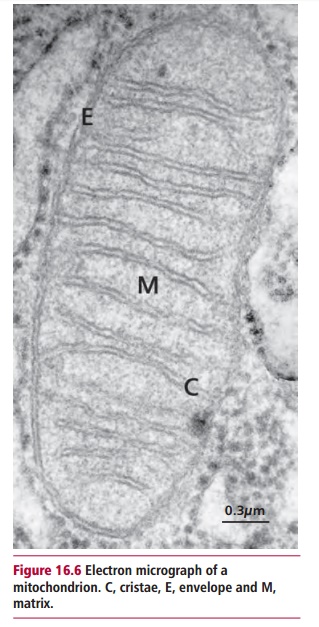
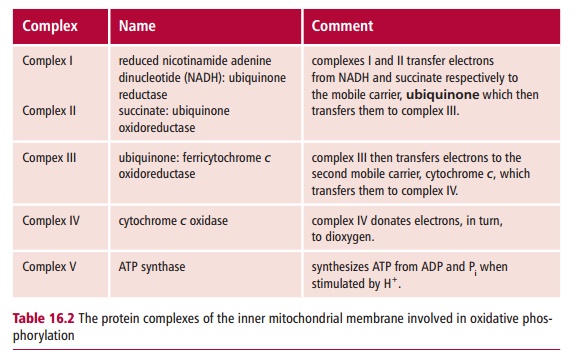
Mitochondria are organelles found in almost all eukaryotic cells although numbers vary from one to several hundred per cell. They are bounded by a mitochondrial envelope that consists of outer and inner mitochondrial membranes with infoldings called cristae, and encloses a central region called the mitochondrial matrix (Figure 16.6). The inner mitochondrial membrane contains five protein complexes (Table 16.2) that together with cytochrome c perform the energy transduction reactions with the formation of ATP byoxidative phosphorylation. The matrix contains enzymes that catabolize fuel molecules to yield the reduced coenzymes NADH and FADH2 necessary for oxidative phosphorylation, and produces small organic molecules that are the precursors in biosynthetic metabolism. The mitochondria also help in maintaining the intracellular homeostasis of many metabolites and ions, including Ca2+ and H+.
Mitochondria are also involved in cellular responses to environmental tox-ins that can stimulate them to release a potent mixture of enzymes and other proteins that eventually bring about the death of the cell. This is one form of apoptosis or cell suicide. These apoptotic mechanisms are, of course, understrict regulation, but defects in these controls cause some neurodegenerative and autoimmune diseases and some cancers.
Mitochondria are unusual organelles, in that the matrix also contains molecules of mitochondrial DNA (mtDNA) containing mitochondrial genes, in addition to mitochondrial ribosomes and mitochondrial transfer RNA molecules. Mitochondrial ribosomes differ in size and structure from those present in the cytosol.
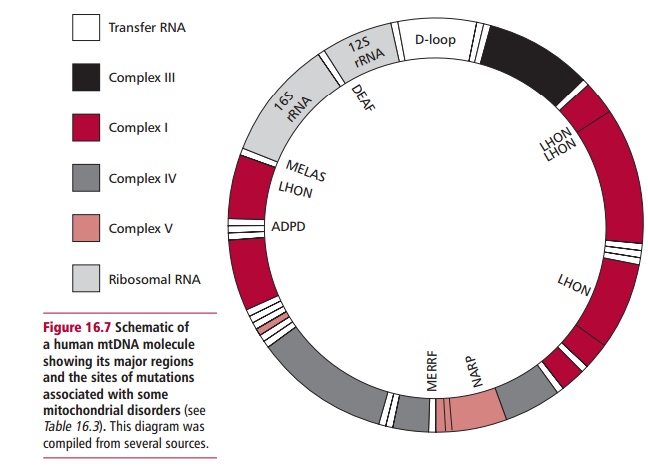
Human mitochondrial DNA is a circular double-stranded molecule of 16 569 base pairs present in multiple copies in each mitochondrion. Human mtDNA has been completely sequenced and contains only 37 genes (Figure 16.7). Twenty-eight genes are encoded on one of the strands of the DNA and nine genes on the other. These genes encode 22 transfer RNAs, the 12S and 16S mitochondrial ribosomal RNAs, and 13 mitochondrial polypeptides that are involved in oxidative phosphorylation. Thus, mitochondria have the genetic information and machinery to synthesize proteins. Mitochondria contain approximately 1000 different proteins, that is about 3% of all cellular proteins. It follows that the majority of mitochondrial proteins are encoded by nuclear genes and are synthesized on ribosomes in the cytosol. These proteins require transportation across one or both of the mitochondrial membranes to reach their sites of activity.
CAUSES OF MITOCHONDRIAL DISORDERS
Mitochondrial disorders are a heterogeneous group of disorders resulting from impairment of the mitochondrial oxidative phosphorylation system.

They are associated not only with numerous mutations of the mtDNA but also mutations in nuclear genes that affect processes such as the assembly of protein subunits and the import of proteins from the cytosol into the mitochondrion. Such mutations can affect the morphology of the mitochondrion (Figure 16.8).
Mitochondria replicate by simple divisions that are independent of mitosis and meiosis. The mitochondrial chromosomes are copied prior to replication. However, the replicating enzyme, mtDNA-dependent DNA polymerase or DNA polymerase F, replicates DNA with much poorer fidelity than the nuclear DNA polymerases. Thus the mutation rates of mitochondrial genes are esti-mated to be about 10 times greater than those of nuclear genes. This may contribute to the aging process . Furthermore, the mitochondrial genome resembles that of bacteria in that the genes lack introns and repeated DNA sequences so that about 93% of the DNA is transcribed as opposed to the 5% or less for the nuclear genome. Thus, despite its relatively small size, a relatively large number of mutations are associated with mtDNA and these mutations are significant contributors to human disease.
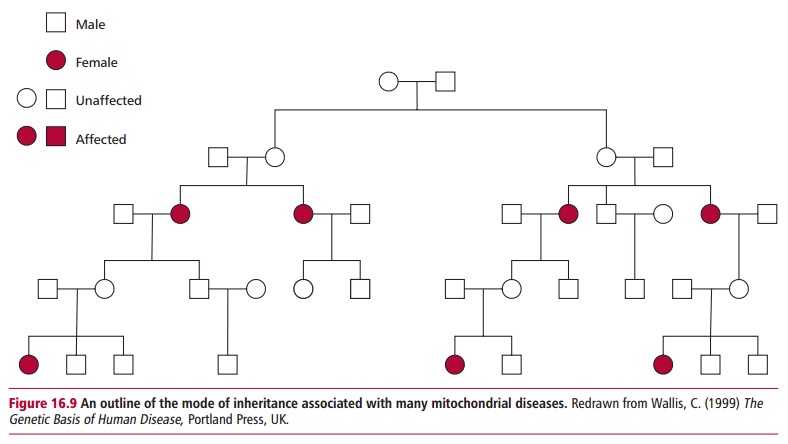
Mitochondrial disorders are caused by similar types of mutations to those that cause diseases associated with the nuclear genome, that is, point muta-tions, deletions, and duplications However, the inheritance of these diseases differs strikingly to that of the nuclear genetic diseases. In zygote formation, a spermatozoon contributes its nuclear genome but not its mitochondrial genome to the egg cell. The resulting fertilized zygote contains only the mitochondria that were present in the unfertilized egg and are there-fore entirely maternal in origin. Thus all children, male and female, inherit their mitochondria only from their mother, and males cannot transmit their mitochondria to subsequent generations (Figure 16.9). Thus a typical disease resulting from a mutation in mtDNA is an inherited condition that can affect both sexes but can only be passed on by affected mothers.
The first disease to be directly linked to mutations in mtDNA was Leber hereditary optic neuropathy (LHON), an inherited neuropathology. This disease presents in midlife with a rapid bilateral loss of central vision due to atrophy of the optic nerve leading to blindness. Early studies of LHON showed a puzzling maternal pattern of inheritance, with more males than females affected. In 1988 it was demonstrated that LHON was caused by mutations in the mtDNA. Three main mutations are present in at least 90% of families with the disease and all cause substitutions of highly conserved amino acid residues in the affected mitochondrial proteins. Thus the mtDNA mutations explain the maternal transmission of LHON although the reason why a higher proportion of males than females are affected is not known. It has been suggested that the expression of the disease could also require the coinheritance of an X-linked recessive mutation; the development of LHON could be hormonally influenced by androgens or by environmental factors, such as heavy tobacco smoking, may also contribute to the progression of the condition.
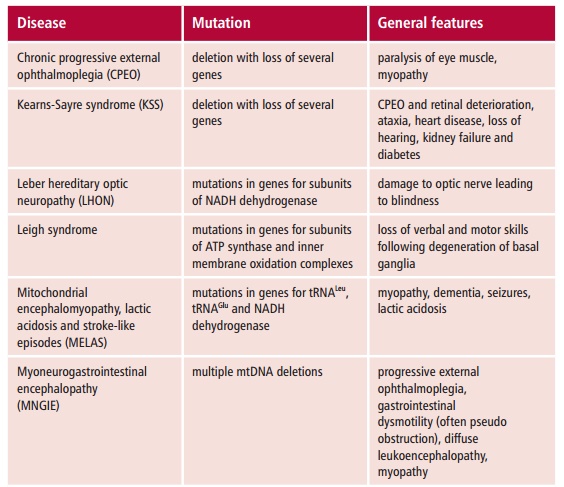
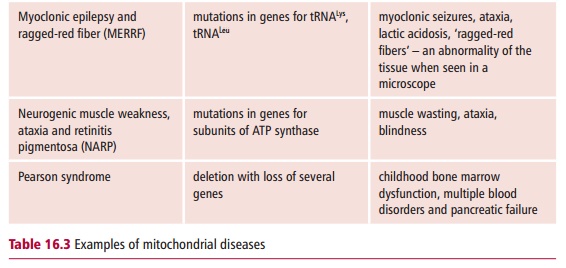
More than 100 pathogenic point mutations and innumerable rearrangements of mtDNA are known that lead to general, multisystem disorders (Table 16.3), which, like LHON, are often neuromuscular in nature. Perhaps not surpris-ingly, most mitochondrial disorders tend to affect the most energy demand-ing tissues, such as the central nervous system, heart and skeletal muscles, kidneys and secretory tissues, although this is not always the case since mutant proteins may interfere with mitochondrial functions in addition to ATP formation. The diseases also vary in severity in patients with the same mtDNA mutation. The term homoplasmy refers to the condition in which every mtDNA molecule has the same causative mutation. In contrast, hetero-plasmy occurs when the cell has a mixed population of normal and mutantmitochondria. If heteroplasmy occurs, the severity and symptoms of the dis-ease may depend on the proportion of abnormal mtDNA in some critical tis-sue. The proportion can also differ between mother and child because of the random segregation of mtDNA molecules at cell division.
Mitochondrial disorders are far more common than was previously thought and a substantial increase in the number of genetic defects have been reported in patients. In the UK, it is estimated that mitochondrial disorders occur with a frequency of about seven in 100 000.
DIAGNOSIS AND TREATMENT OF MITOCHONDRIAL DISORDERS
Considerable advances have been made in our understanding of the pathol-ogy of mitochondrial disorders. However, the diagnosis and detection of mtDNA mutations is problematic because of the varied etiology and clinical features of these diseases (Table 16.3). Similarly, predictors of disease progres-sion are also highly unsatisfactory. The concentrations of lactate in plasma, cerebrospinal fluid and urine may, individually or collectively, be increased relative to normal, although these changes are also seen in numerous other clinical conditions. However, a plasma lactate : pyruvate ratio of greater than 40 is usually considered a significant indicator of mitochondrial dysfunc-tion in adults. Other indicators may be the presence of organic acids and myoglobin in the urine, ketoacidosis , and impaired renal, liver and glandular functions.
It would appear that several different approaches to treating and managing mitochondrial disorders will be necessary. Most current treatments of mitochondrial disorders are unsatisfactory and concentrate merely on relieving symptoms. For example, administration of hydrogen carbonate and dialysis may be used to relieve lactate acidosis. Exercise may decrease the proportion of mutant mtDNA in muscle. Treatment with standard vitamins and ubiquinone supplements has been tried but any benefits have yet to be established.
Gene therapy using allotopic expression may offer a permanent cure in the future. The strategy behind this technique is to insert a mitochondrial gene into the nucleus where it is transcribed. The resulting messenger RNA is trans-lated in the cytosol and the protein product is then imported into the mito-chondria where it alleviates the effects of the mtDNA mutation.
Families affected by mitochondrial disorders should be offered counseling on prognosis, risk of recurrence and prevention but even this is complex, because the degree of mutant mtDNA can change dramatically during each transmission of mitochondria from a mother to a child. The advice to affected families on potential reproductive options may therefore be unsatisfactory. Consequently, the clinical management of patients with mitochondrial disorders is largely supportive.
Related Topics