Chapter: Biology of Disease: Membrane, Organelle and Cytoskeletal Disorders
Peroxisomal Disorders
PEROXISOMAL DISORDERS
Peroxisomes (Figure 16.10) are organelles surrounded by a single membrane, that carry out certain oxidation reactions, particularly those involved in the partial degradation of long chain fatty acids. Unlike mitochondria, these oxidations are not coupled to the formation of ATP. Peroxisomal proteins are synthesized in the cytosol and imported into the organelles by a com-plex process that relies upon the relevant protein possessing a peroxisomal targeting signal (PTS). The major signal (PTS1) consists of the carboxyl ter-minal three amino acid residues being the consensus sequence –Ser-Lys-Leu-COO–.
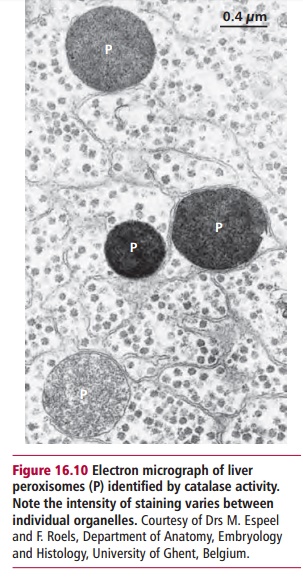
Defects in peroxisomes produce a variety of lethal human diseases. The most common of these are adrenoleukodystrophy (ALD), Zellweger syndrome and Refsum’s disease. These diseases are caused by defects in individual peroxisomal enzymes or faults in the transport system necessary to convey proteins from their site of synthesis in the cytosol to the peroxisome. All are rare, occurring in approximately one in 50 000 live births.
Adrenoleukodystrophy is an X-linked disease caused by muta-tions in a gene encoding an integral peroxisome membrane protein that acts as an active carrier across the peroxisomal membrane. The disease is charac-terized by increased concentrations of very long chain fatty acids (VLCFAs) in the plasma and general tissues and progressive adrenal cortex dysfunc-tion. Several types of ALD occur, the most severe affecting males of four to eight years of age. The major symptoms are learning disabilities, seizures, deafness, dysarthria, dysphagia, poor muscular coordination, vomiting, increased pigmentation of the skin, progressive dementia and poor behav-ior, such as aggression and hyperactivity. The adult form normally presents at 20 to 36 years. Symptoms include stiffness and a progressive paraparesis or weakness of the legs and ataxia, an inability to control muscular move-ments. Progress of the adult form is slower than childhood ALD, but may also result in loss of brain function and may be confused with multiple scle-rosis. Occasionally, women carriers can show mild symptoms of ALD.
Zellweger syndrome is caused by defects in the general ability to import pro-teins into peroxisomes. The peroxisomal enzymes remain in the cytosol and are rapidly degraded leading to a severe peroxisomal deficiency. Individuals have severe abnormalities in their brain, liver and kidneys and die soon after birth. Refsum’s disease or hereditary motor sensory neuropathy type IV, heredopathia atactica polyneuritiformis, was first recognized in the 1940s by Refsum, who observed markedly increased concentrations of phytanic acid, a major constituent of dairy foods, in certain patients. The clinical features include the accumulation of phytanic acid in plasma and lipid-containing tis-sues, retinitis pigmentosa, blindness, anosmia (loss of sense of smell), deaf-ness, sensory neuropathy and ataxia.
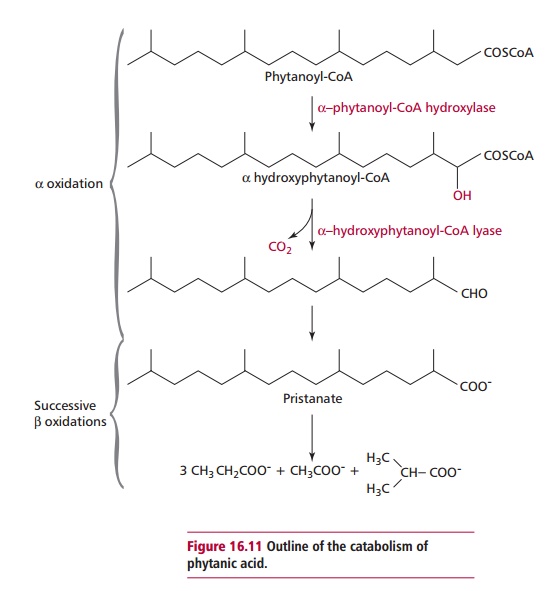
Refsum’s disease is an autosomal recessive disorder that affects the α -oxidation of phytanic acid. In contrast to Zellweger syndrome, Refsum’s disease is due to a reduced enzyme activity. Two enzymes, @-phytanoyl-CoA hydroxylase (PAHX) and α -hydroxyphytanoyl-CoA lyase, found in peroxisomes, are necessary for the oxidation of phytanic acid (Figure 16.11). Mutant forms of PAHX have been shown to be responsible for some cases of Refsum’s disease. An infantile form of Refsum’s disease also leads to an accumulation of phytanic acid but resembles Zellweger syndrome in that it is due to general defects in the transport of appropriate peroxisomal enzymes into the organelle. It is relatively less severe than Zellweger syndrome because the enzymes in the cytosol are thought to have a longer half-life.
DIAGNOSIS AND TREATMENT OF PEROXISOMAL DISORDERS
The diagnosis of peroxisomal disorders usually involves biochemical tests, such as determining the concentrations of VLCFAs in the plasma. Observations of deficient secretions of aldosterone and cortisol by the adrenal cortex can imply ALD. Prenatal diagnoses are available using cultured amniocytes and chorionic villus cells
The treatment of patients with peroxisomal defects is problematic since many, especially those with Zellweger syndrome, have significant brain damage at birth so a full recovery is not possible even with postnatal therapy. Patients with some of the relatively milder conditions can live into their second decade.
Treatment largely involves supportive care and symptomatic therapy. Patients with ALD may be given Lorenzo’s oil (glyceroyl trioleate and trierucate oils) and a strict dietary regimen that returns VLCFAs to normal concentrations but does not prevent neurological degeneration. A bone marrow transplant usually has a successful outcome but only if diagnosis is early and the donor is a perfect HLA match . The treatment of Zellweger syndrome is limited. The most beneficial is the administration of the essential long chain fatty acid, docosahexaenoic acid . Sufferers of Refsum’s disease are given a dietary regimen free of phytanic acid.
Related Topics