Chapter: Biology of Disease: Membrane, Organelle and Cytoskeletal Disorders
Causes of Mitochondrial Disorders
CAUSES OF MITOCHONDRIAL DISORDERS
Mitochondrial disorders are a heterogeneous group of disorders resulting from impairment of the mitochondrial oxidative phosphorylation system.
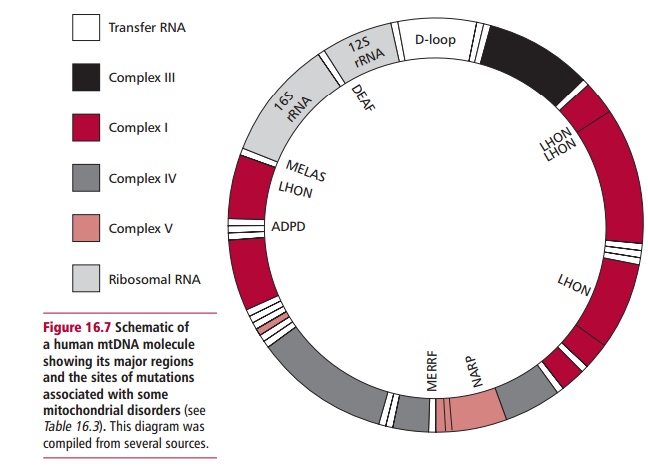

They are associated not only with numerous mutations of the mtDNA but also mutations in nuclear genes that affect processes such as the assembly of protein subunits and the import of proteins from the cytosol into the mitochondrion. Such mutations can affect the morphology of the mitochondrion (Figure 16.8).
Mitochondria replicate by simple divisions that are independent of mitosis and meiosis. The mitochondrial chromosomes are copied prior to replication. However, the replicating enzyme, mtDNA-dependent DNA polymerase or DNA polymerase F, replicates DNA with much poorer fidelity than the nuclear DNA polymerases. Thus the mutation rates of mitochondrial genes are esti-mated to be about 10 times greater than those of nuclear genes. This may contribute to the aging process . Furthermore, the mitochondrial genome resembles that of bacteria in that the genes lack introns and repeated DNA sequences so that about 93% of the DNA is transcribed as opposed to the 5% or less for the nuclear genome. Thus, despite its relatively small size, a relatively large number of mutations are associated with mtDNA and these mutations are significant contributors to human disease.
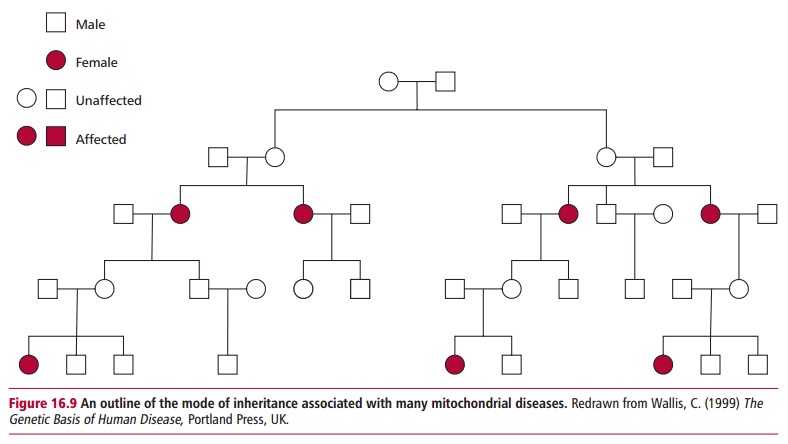
Mitochondrial disorders are caused by similar types of mutations to those that cause diseases associated with the nuclear genome, that is, point muta-tions, deletions, and duplications However, the inheritance of these diseases differs strikingly to that of the nuclear genetic diseases. In zygote formation, a spermatozoon contributes its nuclear genome but not its mitochondrial genome to the egg cell. The resulting fertilized zygote contains only the mitochondria that were present in the unfertilized egg and are there-fore entirely maternal in origin. Thus all children, male and female, inherit their mitochondria only from their mother, and males cannot transmit their mitochondria to subsequent generations (Figure 16.9). Thus a typical disease resulting from a mutation in mtDNA is an inherited condition that can affect both sexes but can only be passed on by affected mothers.
The first disease to be directly linked to mutations in mtDNA was Leber hereditary optic neuropathy (LHON), an inherited neuropathology. This disease presents in midlife with a rapid bilateral loss of central vision due to atrophy of the optic nerve leading to blindness. Early studies of LHON showed a puzzling maternal pattern of inheritance, with more males than females affected. In 1988 it was demonstrated that LHON was caused by mutations in the mtDNA. Three main mutations are present in at least 90% of families with the disease and all cause substitutions of highly conserved amino acid residues in the affected mitochondrial proteins. Thus the mtDNA mutations explain the maternal transmission of LHON although the reason why a higher proportion of males than females are affected is not known. It has been suggested that the expression of the disease could also require the coinheritance of an X-linked recessive mutation; the development of LHON could be hormonally influenced by androgens or by environmental factors, such as heavy tobacco smoking, may also contribute to the progression of the condition.
More than 100 pathogenic point mutations and innumerable rearrangements of mtDNA are known that lead to general, multisystem disorders (Table 16.3), which, like LHON, are often neuromuscular in nature. Perhaps not surpris-ingly, most mitochondrial disorders tend to affect the most energy demand-ing tissues, such as the central nervous system, heart and skeletal muscles, kidneys and secretory tissues, although this is not always the case since mutant proteins may interfere with mitochondrial functions in addition to ATP formation. The diseases also vary in severity in patients with the same mtDNA mutation. The term homoplasmy refers to the condition in which every mtDNA molecule has the same causative mutation. In contrast, hetero-plasmy occurs when the cell has a mixed population of normal and mutantmitochondria. If heteroplasmy occurs, the severity and symptoms of the dis-ease may depend on the proportion of abnormal mtDNA in some critical tis-sue. The proportion can also differ between mother and child because of the random segregation of mtDNA molecules at cell division.
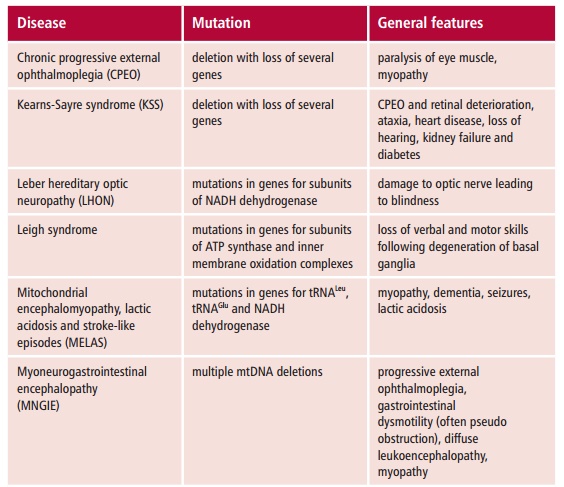
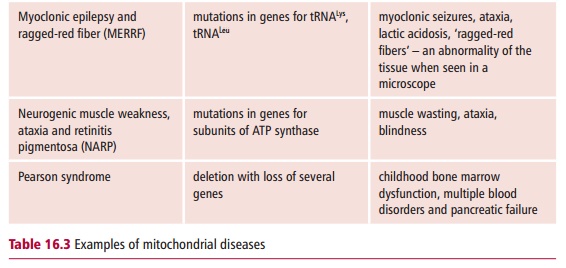
Mitochondrial disorders are far more common than was previously thought and a substantial increase in the number of genetic defects have been reported in patients. In the UK, it is estimated that mitochondrial disorders occur with a frequency of about seven in 100 000.
Related Topics