Chapter: Biology of Disease: Membrane, Organelle and Cytoskeletal Disorders
Lysosomal Disorders
LYSOSOMAL DISORDERS
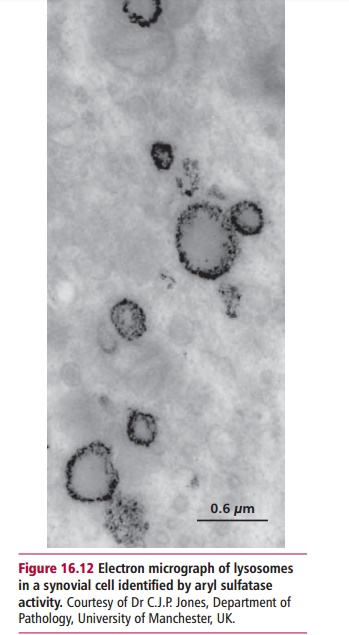
Lysosomes are organelles bounded by a single, impermeable membrane. They contain about 60 different hydrolytic enzymes (hydrolases) that are active at acid pH and are able to degrade all biological materials (Figure 16.12). Other membrane-bound vesicles, endosomes and phagosomes, fuse with lysosomes and deliver materials taken up by the cell by endocytosis or phagocytosis for degradation by these enzymes.
Lysosomal enzymes are synthesized by the ribosomes of the rough endoplasmic reticulum and enzymes within the Golgi apparatus. In the Golgi apparatus, synthetic enzymes recognize sites on the surface of the prolysosomal enzymes and tag them with mannose 6-phosphate residues that act as molecular addresses, ensuring that they are integrated into vesicles that eventually become lysosomes.
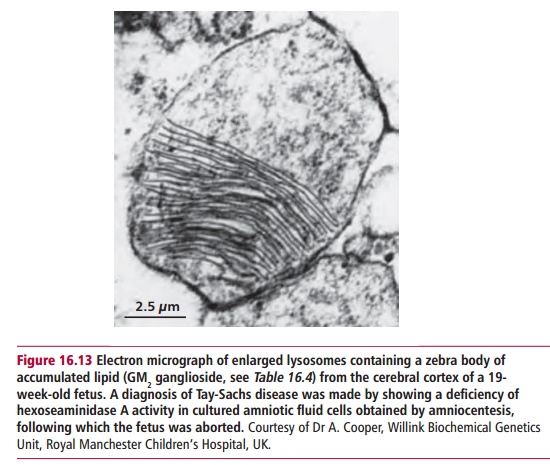
Lysosomal storage diseases (LSDs) are caused by mutations in genes encoding lysosomal hydrolases leading to an accumulation of the substrate for the inactive enzyme within the lysosome (Figure 16.13). Eventually the cell dies, impairing the functions of major organs such as the brain and liver. More than 40 lysosomal storage diseases are known (Table 16.4). All are relatively uncommon in the general population although several are more prevalent among Ashkenazi Jews.
Lysosomal storage diseases are congenital disorders that seriously reduce the quality of life and life expectancy. For example, Tay-Sachs disease leads to blindness, paralysis, dementia and death, usually by two years of age. In most cases LSDs are caused by the deficiency of a single enzyme. The diseases can be subdivided according to which pathway is affected. For example, enzyme deficiencies in the pathway for degrading glycosaminoglycans cause mucopolysaccharidoses, deficiencies affecting glycopeptides cause glycoproteinosis and glycolipid storage diseases result from deficiencies in enzymes involved in the degradation of sphingolipids.
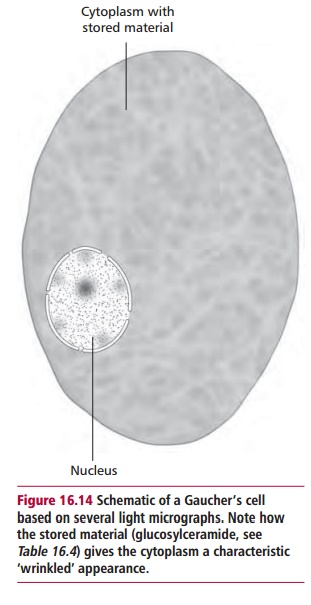
The commonest lysosomal storage disease is Gaucher’s disease with an inci-dence of one in 30 000 to one in 50 000 although it is about 30 times more common in Ashkenazi Jews. The disease is characterized by the presence of histiocytes, cells belonging either to the macrophage or Langerhans cell line-age , enlarged with accumulated lipid. These cells are known as Gaucher’s cells (Figure 16.14) and are found in the bone marrow, reticuloen-dothelial system, parts of the circulation and other organs. The disease is an autosomal inherited recessive condition leading to a deficiency of lysosomal A-glucocerebrosidase and the accumulation of glucosylceramide within lyso-somes. The pathogenic mechanisms are poorly understood but three forms are recognized: type 1 or adult, type 2 or infantile and type 3 or juvenile; the differences presumably arise from different mutations in the gene encoding the enzyme.
Adult, type 1, Gaucher’s disease may present from infancy to adulthood. Although some sufferers may die at a young age due to pulmonary infections, most survive to late adult life. Type 1 disease is characterized by hepatosplenomegaly, the enlargement of the liver typically occurring after that of the spleen. The abdomen can also enlarge due to distention of the colon. Most of these visceral features arise from macrophage dysfunction. The skin bruises very easily and becomes pale yellow in color. Febrile episodes with generalized pain in the body and limbs occur. Painful bones, joints, degeneration of vertebral bodies and arthritic hip joints are common. Pathological fractures may also occur. Mild anemia and recurrent pulmonary infections are characteristic. Some patients show neurological abnormalities, decreased intelligence, with ataxia and fits.
Infantile, type 2, Gaucher’s disease is much more severe with death occur-ring by the age of two years following progressive psychomotor regression. The earliest symptoms include a protuberant abdomen usually due to hepat-osplenomegaly and a general failure to thrive. Indeed, after six months devel-opmental arrest and neurological regression are obvious characteristics. Fits may occur. The patient becomes spastic, with laryngeal spasms causing dif-ficulties in swallowing, and strabismus(a squint or abnormal alignment of the eyes). Deterioration is rapid, leading to recurrent pulmonary infections and death.
Juvenile, type 3, Gaucher’s disease is the rarest of the three. Although it presents later than type 2 and has a slower progression it also has a characteristic neurological deterioration. The major features are hepatosplenomegaly, convulsions and psychomotor retardation. Ataxia, spasticity and strabismus may also occur.
Inclusion cell (I-cell) disease or mucolipidosis II is rare, with an incidence of about one in 640 000. All lysosomal hydrolases are actually produced but a lack of N-acetylglucosylaminyl-1-phosphotransferase activity means they fail to be tagged with mannose 6-phosphate residues, with the result that they are not delivered to lysosomes but are secreted from the cell. The disease is named from the densely staining intracytoplasmic regions present in the fibroblasts of sufferers (Figure 16.15). Overt symptoms are present at birth or appear within the first few years of life. There is psychomotor developmental delay
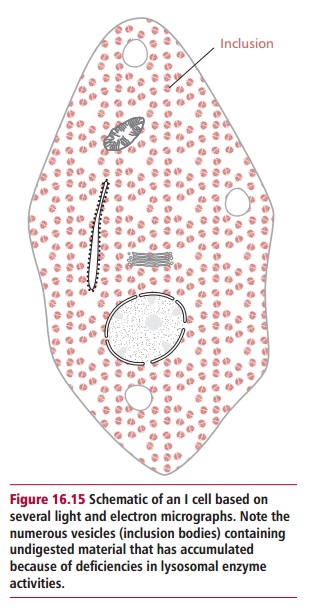
and generally failure to thrive. Hernias, hepatomegaly, joint limitation and hip dislocations are common. Patients experience frequent upper respiratory tract infections with recurrent attacks of pneumonia, bronchitis and otitis media . Cardiac murmurs from pathologies of heart valves are common and survival beyond the age of five years is unusual.
DIAGNOSIS AND TREATMENT OF LYSOSOMAL STORAGE DISORDERS
Experienced laboratory practitioners using relatively simple methods can accurately diagnose patients exhibiting the typical traits of LSDs. However, atypical patients usually require more detailed investigations. An early diagnosis is important to prevent serious damage to the nervous and skeletal systems. Almost all lysosomal storage diseases can be diagnosed using samples of leukocytes or plasma. Diagnosis is based upon assays that determine the reduced activities of lysosomal enzymes or an increase in substrate in skin fibroblasts, plasma or urine. For example, Gaucher’s and I-cell diseases can be confirmed by demonstrating deficiencies in β -glucocerebrosidase and N-acetylglucosylaminyl-1-phosphotransferase activities in leukocytes orfibroblasts respectively.
The prognosis for adult Gaucher’s disease patients is relatively good. The hypersplenism is usually relieved by splenectomy although this may has-ten bone deterioration. Symptomatic treatment includes analgesics for the bone pains, blood transfusions to relieve the anemia and orthopedic relief for fractures and degeneration of the hip joints. Enzyme replacement therapy, involving intravenous administration of purified A-glucocerebrosidase, is now common. Receptors on the surfaces of macrophages selectively bind man-nose residues on the enzyme and allow it to be absorbed by the cells. Within the macrophages, the enzyme is delivered to the lysosomes where it cata-lyzes the breakdown of the accumulated glucosylceramide. In the majority of cases this is an effective and safe treatment, reducing the sizes of the liver and spleen and allowing them and the bone marrow to function effectively. Again, bone marrow transplants may be useful. Gene therapy, in which a functional gene for A-glucocerebrosidase is inserted into stem cells in the bone marrow, may in the future provide a complete cure.
Treatment for I-cell disease is limited. Bone marrow transplants can potentially replace the defective hemopoietic system with stem cells from a healthy donor, providing a replacement for the defective enzymes. In a limited number of cases, bone marrow transplants have normalized lysosomal enzyme activities, but the preexisting clinical damage from the disease is usually so extensive by this stage that this option is of questionable long-term benefit. In addition, bone marrow transplants can fail and the problems of finding compatible donors are immense.
Genetic counseling and prenatal diagnosis have roles in relation to LSDs. In cases where the likelihood of a lysosomal storage disease being present in a fetus is high, prenatal diagnosis is possible and a therapeutic abortion can be offered. Methods for screening all newborns without a family history of such conditions have been proposed but such screening programs would require a large amount of effort for comparatively little gain.
Related Topics