Chapter: Basic & Clinical Pharmacology : Opioid Analgesics & Antagonists
Pharmacokinetics - Basic Pharmacology of the Opioid Analgesics
Pharmacokinetics
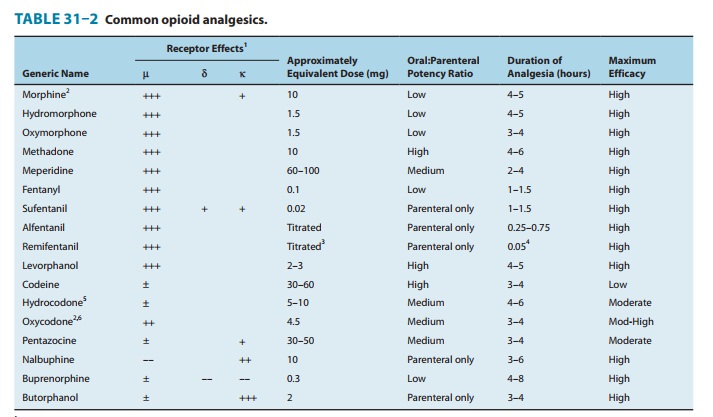
Some properties of
clinically important opioids are summarized in Table 31–2.
A. Absorption
Most opioid analgesics
are well absorbed when given by subcuta-neous, intramuscular, and oral routes.
However, because of the first-pass effect, the oral dose of the opioid (eg,
morphine) may need to be much higher than the parenteral dose to elicit a
thera-peutic effect. Considerable interpatient variability exists in first-pass
opioid metabolism, making prediction of an effective oral dose difficult.
Certain analgesics such as codeine and oxycodone are effective orally because
they have reduced first-pass metabo-lism. Nasal insufflation of certain opioids
can result in rapid therapeutic blood levels by avoiding first-pass metabolism.
Other routes of opioid administration include oral mucosa via lozenges, and
transdermal via transdermal patches. The latter can provide delivery of potent
analgesics over days. Recently an iontophoretic transdermal system has been
introduced, allowing needle-free delivery of fentanyl for patient-controlled
analgesia.
B. Distribution
The uptake of opioids by various organs and tissues is a function of both physiologic and chemical factors. Although all opioids bind to plasma proteins with varying affinity, the drugs rapidly leave the blood compartment and localize in highest concentra-tions in tissues that are highly perfused such as the brain, lungs, liver, kidneys, and spleen. Drug concentrations in skeletal muscle may be much lower, but this tissue serves as the main reservoir because of its greater bulk. Even though blood flow to fatty tissue is much lower than to the highly perfused tissues, accumulation can be very important, particularly after frequent high-dose administration or continuous infusion of highly lipophilic opioids that are slowly metabolized, eg, fentanyl.
C. Metabolism
The opioids are converted
in large part to polar metabolites (mostly glucuronides), which are then
readily excreted by the kidneys. For example, morphine, which contains free
hydroxyl groups, is primarily conjugated to morphine-3-glucuronide (M3G), a
compound with neuroexcitatory properties. The neuro-excitatory effects of M3G do
not appear to be mediated by receptors but rather by the GABA/glycinergic
system. In con-trast, approximately 10% of morphine is metabolized to
morphine-6-glucuronide (M6G), an active metabolite with analgesic potency four
to six times that of its parent compound. However, these relatively polar
metabolites have limited ability to cross the blood-brain barrier and probably
do not contribute significantly to the usual CNS effects of morphine given acutely.
Nevertheless, accu-mulation of these metabolites may produce unexpected adverse
effects in patients with renal failure or when exceptionally large doses of
morphine are administered or high doses are administered over long periods.
This can result in M3G-induced CNS excita-tion (seizures) or enhanced and
prolonged opioid action producedby M6G. CNS uptake of M3G and, to a lesser
extent, M6G can be enhanced by co-administration with probenecid or with drugs
that inhibit the P-glycoprotein drug transporter. Like morphine, hydromorphone
is metabolized by conjugation, yielding hydro-morphone-3-glucuronide (H3G),
which has CNS excitatory properties. However, hydromorphone has not been shown
to form significant amounts of a 6-glucuronide metabolite.
The effects of these
active metabolites should be considered in patients with renal impairment
before the administration of mor-phine or hydromorphone, especially when given
at high doses.
Esters (eg, heroin,
remifentanil) are rapidly hydrolyzed by common tissue esterases. Heroin
(diacetylmorphine) is hydrolyzed to monoacetylmorphine and finally to morphine,
which is then conjugated with glucuronic acid.
Hepatic oxidative
metabolism is the primary route of degrada-tion of the phenylpiperidine opioids
(meperidine, fentanyl, alfen-tanil, sufentanil) and eventually leaves only
small quantities of the parent compound unchanged for excretion. However,
accumula-tion of a demethylated metabolite of meperidine, normeperidine, may
occur in patients with decreased renal function and in those receiving multiple
high doses of the drug. In high concentrations, normeperidine may cause
seizures. In contrast, no active metabo-lites of fentanyl have been reported.
The P450 isozyme CYP3A4 metabolizes fentanyl by N-dealkylation in the liver. CYP3A4 is also present in the mucosa
of the small intestine and contributes to the first-pass metabolism of fentanyl
when it is taken orally. Codeine, oxycodone, and hydrocodone undergo metabolism
in the liver by P450 isozyme CYP2D6, resulting in the production of metabolites
of greater potency. For example, codeine is de-methylated to morphine. Genetic
polymorphism of CYP2D6 has been documented and linked to the variation in
analgesic response seen among patients. Nevertheless, the metabolites of oxycodone
and hydrocodone may be of minor consequence; the parent com-pounds are
currently believed to be directly responsible for the majority of their
analgesic actions. However, oxycodone and its metabolites can accumulate under
conditions of renal failure and have been associated with prolonged action and
sedation. In the case of codeine, conversion to morphine may be of greater
impor-tance because codeine itself has relatively low affinity for opioid
receptors. As a result, patients may experience either no significant analgesic
effect or an exaggerated response based on differences in metabolic conversion.
For this reason, routine use of codeine, especially in pediatric age groups, is
being reconsidered.
D. Excretion
Polar metabolites,
including glucuronide conjugates of opioid anal-gesics, are excreted mainly in
the urine. Small amounts of unchanged drug may also be found in the urine. In
addition, glucuronide con-jugates are found in the bile, but enterohepatic
circulation repre-sents only a small portion of the excretory process.
Related Topics