Chapter: Basic & Clinical Pharmacology : Opioid Analgesics & Antagonists
Pharmacodynamics - Basic Pharmacology of the Opioid Analgesics
Pharmacodynamics
A. Mechanism of Action
Opioid agonists
produce analgesia by binding to specific G pro-tein-coupled receptors that are
located in brain and spinal cord regions involved in the transmission and
modulation of pain (Figure 31–1). Some effects may be mediated by opioid
receptors on peripheral sensory nerve endings.
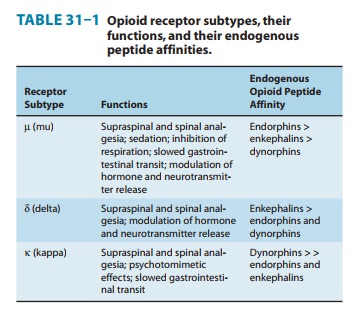
1. Receptor types—As noted previously,
three major classes ofopioid receptors (μ, δ, and κ) have been identified in various ner-vous
system sites and in other tissues (Table 31–1). Each of the three major
receptors has now been cloned. All are members of the G protein-coupled family
of receptors and show significant amino acidsequence homologies. Multiple
receptor subtypes have been pro-posed based on pharmacologic criteria,
including μ1, μ2; δ1, δ2; and κ1, κ2, and κ3. However, genes encoding only one subtype
fromeach of the μ,
δ,
and κ
receptor families have been isolated and characterized thus far. One plausible
explanation is that μ-receptor
subtypes arise from alternate splice variants of a common gene. This idea has
been supported by the identification of receptor splice vari-ants in mice and
humans. Since an opioid may function with differ-ent potencies as an agonist,
partial agonist, or antagonist at more than one receptor class or subtype, it
is not surprising that these agents are capable of diverse pharmacologic
effects.
2. Cellular actions— At the molecular
level, opioid receptorsform a family of proteins that physically couple to G
proteins andthrough this interaction affect ion channel gating, modulate
intra-cellular Ca2+ disposition, and alter protein phosphorylation . The opioids have two
well-established direct G protein-coupled actions on neurons: (1) they close
voltage-gated Ca2+ channels on presynaptic nerve terminals and thereby reduce
transmitter release, and (2) they hyperpolarize and thus inhibit postsynaptic
neurons by opening K+ channels. Figure 31–1 schematically illustrates these effects.
The presynaptic action— depressed transmitter release—has been demonstrated for
release of a large number of neurotransmitters including gluta-mate, the
principal excitatory amino acid released from nocicep-tive nerve terminals, as
well as acetylcholine, norepinephrine, serotonin, and substance P.
3. Relation of physiologic effects
to receptor type—Themajority of currently available opioid analgesics act
primarily at the μ-opioid
receptor (Table 31–2). Analgesia and the euphoriant, respiratory depressant,
and physical dependence properties of morphine result principally from actions
at μ
receptors. In fact, the μ receptor was originally defined using the
relative potencies for clinical analgesia of a series of opioid alkaloids.
However, opi-oid analgesic effects are complex and include interaction with δ and κ receptors. This is
supported by the study of genetic knock-outs of the μ, δ, and κ genes in mice. Delta-receptor agonists retain
analgesic properties in δ receptor knockout mice. The development of μ-receptor–selective
agonists could be clinically useful if their side-effect profiles (respiratory
depression, risk ofdependence) were more favorable than those found with current
μ-receptor
agonists, such as morphine. Although morphine doesact at κ and δ receptor sites, it is
unclear to what extent this con-tributes to its analgesic action. The
endogenous opioid peptides differ from most of the alkaloids in their affinity
for the δ
and κ receptors
(Table 31–1).
In an effort to
develop opioid analgesics with a reduced
incidence of respiratory depression or propensity for addiction and
dependence, compounds that show preference for κ opioid recep-tors have been developed. Butorphanol
and nalbuphine have shown some clinical success as analgesics, but they can
cause dys-phoric reactions and have limited potency. It is interesting that
butorphanol has also been shown to cause significantly greater analgesia in
women than in men. In fact, gender-based differences in analgesia mediated by μ- and δ-receptor activation
have been widely reported.
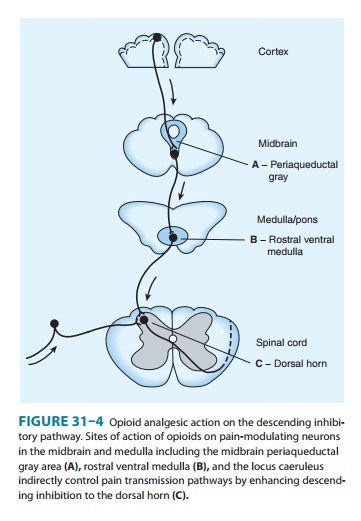
4. Receptor distribution and neural mechanisms of analgesia—Opioid receptor binding sites have been localizedautoradiographically with high-affinity radioligands and with anti-bodies to unique peptide sequences in each receptor subtype. All three major receptors are present in high concentrations in the dorsal horn of the spinal cord. Receptors are present both on spinal cord pain transmission neurons and on the primary afferents that relay the pain message to them (Figure 31–2, sites A and B). Although opioid agonists directly inhibit the dorsal horn pain transmission neurons, they also inhibit the release of excitatorytransmitters from the primary afferents. Within the presynaptic terminals, there is evidence that heterodimerization of the μ-opioid and δ-opioid receptors contribute to μ-agonist efficacy (eg, inhibi-tion of presynaptic voltage-gated calcium channel activity).
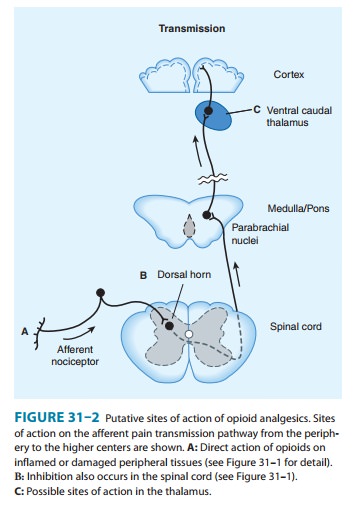
On the other hand, a recent study using a transgenic mouse that expresses a δ–receptor-enhanced green fluorescent protein (eGFP) fusion protein shows little overlap of μ receptor and δ receptor in the dorsal root ganglion neurons. Importantly, the μ receptor is associatedwith TRPV1 and peptide (substance P)-expressing nociceptors, whereas δ-receptor expression predominates in the non-peptidergic population of nociceptors, including many primary afferents with myelinated axons. This is consistent with the action of intrathecal μ-receptor– and δ-receptor–selective ligands that are found to blockheat versus mechanical pain processing, respectively. To what extent the differential expression of the μ receptor and δ receptor in the dorsal root ganglia is characteristic of neurons throughout the CNS remains to be determined.
Thus, opioids exert a
powerful analgesic effect directly on the spinal cord. This spinal action has been exploited
clinically by direct application of opioid agonists to the spinal cord, which
provides a regional analgesic effect while reducing the unwanted respiratory
depression, nausea and vomiting, and sedation that may occur from the supraspinal actions of systemically
adminis-tered opioids.
Under most
circumstances, opioids are given systemically and so act simultaneously at
multiple sites. These include not only the ascending pathways of pain
transmission beginning with specialized peripheral sensory terminals that
transduce painful stimuli (Figure 31–2) but also descending (modulatory)
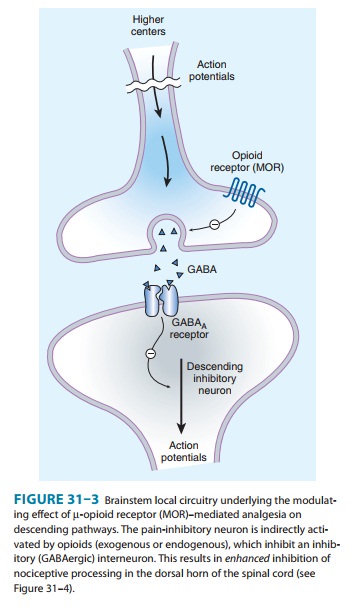
path-ways (Figure 31–3). At these sites as at others, opioids directly inhibit
neurons; yet this action results in the activation
of descend-ing inhibitory neurons that send processes to the spinal cord and
inhibit pain transmission neurons. This activation has been shown to result
from the inhibition of inhibitory neurons in several loca-tions (Figure 31–4).
Taken together, interactions at these sites increase the overall analgesic
effect of opioid agonists.
When pain-relieving
opioid drugs are given systemically, they presumably act upon neuronal circuits
normally regulated by endogenous opioid peptides. Part of the pain-relieving
action of exogenous opioids involves the release of endogenous opioid peptides.
An exogenous opioid agonist (eg, morphine) may act primarily and directly at
the μ
receptor, but this action may evoke the release of endogenous opioids that
additionally act at and κ receptors. Thus, even a receptor-selective
ligand can initi-ate a complex sequence of events involving multiple synapses,
transmitters, and receptor types.
Animal and human
clinical studies demonstrate that both endogenous and exogenous opioids can
also produce opioid-mediated analgesia at sites outside the CNS. Pain associated with inflammation seems especially
sensitive to these peripheral opioid actions. The presence of functional μ receptors on the
peripheral terminals of sensory neurons supports this hypothesis.
Furthermore,
activation of peripheral μ receptors results in a decrease in sensory
neuron activity and transmitter release. The endogenous release of β-endorphin produced by
immune cells within injured or inflamed tissue represents one source of
physi-ologic peripheral μ-receptor activation. Peripheral
administration of opioids, eg, into the knees of patients following
arthroscopic knee surgery, has shown clinical benefit up to 24 hours after
administration. If they can be developed, opioids selective for a peripheral
site would be useful adjuncts in the treatment of inflammatory pain (see Box:
Ion Channels & Novel Analgesic Targets). Such compounds could have the
additional benefit of reducing unwanted effects such as constipation.
5. Tolerance
and dependence— With frequently repeatedtherapeutic doses of morphine or its
surrogates, there is a gradual loss in effectiveness; this loss of effectiveness
is denoted tolerance. To reproduce the original response, a larger dose must be
admin-istered. Along with tolerance, physical dependence develops. Physical
dependence is defined as a characteristic withdrawal
or abstinence syndrome when a drug
is stopped or an antagonist isadministered.
The mechanism of
development of tolerance and physical dependence is poorly understood, but
persistent activation ofreceptors such as occurs with the treatment of severe
chronic pain appears to play a primary role in its induction and mainte-nance.
Current concepts have shifted away from tolerance being driven by a simple
up-regulation of the cyclic adenosine mono-phosphate (cAMP) system. Although
this process is associated with tolerance, it is not sufficient to explain it.
A second hypoth-esis for the development of opioid tolerance and dependence is
based on the concept of receptor
recycling. Normally, activation of μ receptors by endogenous ligands results in
endocytosis fol-lowed by resensitization and recycling of the receptor to the
plasma membrane . However, using geneticallymodified mice, research now shows
that the failure of morphine to
induce endocytosis of the μ-opioid receptor is an important com-ponent of
tolerance and dependence. In contrast, methadone, a μ-receptor agonist used for thetreatmentof opioid tolerance
anddependence, does induce receptor endocytosis. This suggests that maintenance
of normal sensitivity of μ receptors requires reactiva-tion by
endocytosis and recycling. Another area of research sug-gests that the δ opioid receptor
functions as an independent component in the maintenance of tolerance. In
addition, the con-cept of receptor
uncoupling has gained prominence. Under this hypothesis, tolerance is due
to a dysfunction of structural interac-tions between the μ receptor and G
proteins, second-messenger systems, and their target ion channels. Uncoupling
and recoupling of μ
receptor function is likely linked to receptor recycling. Moreover, the
NMDA-receptor ion channel complex has been shown to play a critical role in
tolerance development and main-tenance because NMDA-receptor antagonists such
as ketamine can block tolerance development. Although a role in endocytosis is
not yet clearly defined, the development of novel NMDA-receptor antagonists or
other strategies to recouple μ receptors to their target ion channels
provides hope for achieving a clinically effective means to prevent or reverse
opioid analgesic tolerance.
In addition to the
development of tolerance, persistent admin-istration of opioid analgesics has
been observed to increase the
sensation of pain leading to a state of hyperalgesia. This phenom-enon has been
observed with several opioid analgesics, including morphine, fentanyl, and
remifentanil. Spinal dynorphin and acti-vation of the bradykinin receptor have
emerged as important candidates for the mediation of opioid-induced
hyperalgesia.
B. Organ System Effects of Morphine and Its Surrogates
The actions described
below for morphine, the prototypic opioid agonist, can also be observed with
other opioid agonists, partial agonists, and those with mixed receptor effects.
Characteristics of specific members of these groups are discussed below.
1. Central nervous system
effects—The
principal effects ofopioid analgesics with affinity for μ receptors are on the
CNS; the more important ones include analgesia, euphoria, sedation, and
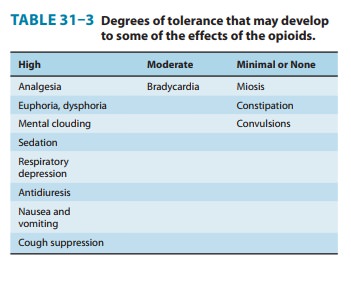
respiratory depression. With repeated use, a high degree of toler-ance occurs
to all of these effects (Table 31–3).
a. Analgesia—Pain consists of both sensory and affective (emo-tional) components. Opioid analgesics are unique in that they can reduce both aspects of the pain experience, especially the affective aspect. In contrast, nonsteroidal anti-inflammatory analgesic drugs have no significant effect on the emotional aspects of pain.
b. Euphoria—Typically, patients or
intravenous drug users whoreceive intravenous morphine experience a pleasant
floating sensa-tion with lessened anxiety and distress. However, dysphoria, an
unpleasant state characterized by restlessness and malaise, may sometimes
occur.
c. Sedation—Drowsiness and clouding of mentation are com-mon effects of opioids. There is little or no amnesia. Sleep isinduced by opioids more frequently in the elderly than in young, healthy individuals. Ordinarily, the patient can be easily aroused from this sleep. However, the combination of morphine with other central depressant drugs such as the sedative-hypnotics may result in very deep sleep. Marked sedation occurs more frequently with compounds closely related to the phenanthrene derivatives and less frequently with the synthetic agents such as meperidine and fentanyl. In standard analgesic doses, morphine (a phenan-threne) disrupts normal rapid eye movement (REM) and non-REM sleep patterns. This disrupting effect is probably characteristic of all opioids. In contrast to humans, a number of species (cats, horses, cows, pigs) may manifest excitation rather than sedation when given opioids. These paradoxic effects are at least partially dose-dependent.
d. Respiratory depression—All of the opioid
analgesics canproduce significant respiratory depression by inhibiting
brain-stem respiratory mechanisms. Alveolar PCO2 may increase, but the most reliable indicator
of this depression is a depressed response to a carbon dioxide challenge. The
respiratory depres-sion is dose-related and is influenced significantly by the
degree of sensory input occurring at the time. For example, it is possible to
partially overcome opioid-induced respiratory depression by stimulation of
various sorts. When strongly painful stimuli that have prevented the depressant
action of a large dose of an opioid are relieved, respiratory depression may
suddenly become marked.
A small to moderate
decrease in respiratory function, as measured by PaCO2 elevation, may be well tolerated in the
patient without prior respiratory impairment. However, in individuals with
increased intracranial pressure, asthma, chronic obstructive pul-monary
disease, or cor pulmonale, this decrease in respiratory function may not be
tolerated. Opioid-induced respiratory depression remains one of the most
difficult clinical challenges in the treatment of severe pain. Research is
ongoing to understand and develop analgesic agents and adjuncts that avoid this
effect. Research to overcome this problem is focused on μ-receptor
phar-macology and serotonin signaling pathways in the brainstem respiratory
control centers.
e. Cough suppression—Suppression of the
cough reflex is awell-recognized action of opioids. Codeine in particular has
been used to advantage in persons suffering from pathologic cough and in
patients in whom it is necessary to maintain ventilation via an endotracheal
tube. However, cough suppression by opioids may allow accumulation of
secretions and thus lead to airway obstruc-tion and atelectasis.
f. Miosis—Constriction of the
pupils is seen with virtually allopioid agonists. Miosis is a pharmacologic
action to which little or no tolerance develops (Table 31–3); thus, it is
valuable in the diagnosis of opioid overdose. Even in highly tolerant addicts,
mio-sis is seen. This action, which can be blocked by opioid antago-nists, is
mediated by parasympathetic pathways, which, in turn, can be blocked by
atropine.
g. Truncal rigidity—An intensification of tone in the largetrunk muscles has been noted with a number of opioids. It was originally believed that truncal rigidity involved a spinal cord action of these drugs, but there is now evidence that it results from an action at supraspinal levels. Truncal rigidity reduces thoracic compliance and thus interferes with ventilation. The effect is most apparent when high doses of the highly lipid-soluble opioids (eg, fentanyl, sufentanil, alfentanil, remifentanil) are rapidly adminis-tered intravenously. Truncal rigidity may be overcome by admin-istration of an opioid antagonist, which of course will also antagonize the analgesic action of the opioid. Preventing truncal rigidity while preserving analgesia requires the concomitant use of neuromuscular blocking agents.
h. Nausea and
vomiting— The
opioid analgesics can activatethe brainstem chemoreceptor trigger zone to
produce nausea and vomiting. There may also be a vestibular component in this
effect because ambulation seems to increase the incidence of nausea and
vomiting.
I.
the brain. This has
been supported by experiments demonstrating that administration of μ-opioid receptor
agonists such as morphine administered to the anterior hypothalamus produces
hyperthermia, whereas administration of κ agonists induces hypothermia.
2. Peripheral effects
a. Cardiovascular
system—Most
opioids have no significantdirect effects on the heart and, other than
bradycardia, no major effects on cardiac rhythm. Meperidine is an exception to
this gener-alization because its antimuscarinic action can result in
tachycardia. Blood pressure is usually well maintained in subjects receiving
opi-oids unless the cardiovascular system is stressed, in which case
hypotension may occur. This hypotensive effect is probably due to peripheral
arterial and venous dilation, which has been attributed to a number of
mechanisms including central depression of vasomotor-stabilizing mechanisms and
release of histamine. No consistent effect on cardiac output is seen, and the
electrocardiogram is not significantly affected. However, caution should be
exercised in patients with decreased blood volume, because the above mechanisms make these
patients susceptible to hypotension. Opioid analgesics affect cerebral
circulation minimally except when PCO2 rises as a
consequence of respiratory depression. Increased PCO2 leads to cerebral vasodilation associated with a decrease in
cerebral vascular resistance, an increase in cerebral blood flow, and an increase
in intracranial pressure.
b. Gastrointestinal tract—Constipation has long
been recog-nized as an effect of opioids, an effect that does not diminish with
continued use. That is, tolerance does not develop to opioid-induced
constipation (Table 31–3). Opioid receptors exist in high density in the
gastrointestinal tract, and the constipating effects of the opioids are
mediated through an action on the enteric nervous system as well as the CNS. In the stomach, motil-ity
(rhythmic contraction and relaxation) may decrease but tone (persistent
contraction) may increase—particularly in the central portion; gastric
secretion of hydrochloric acid is decreased. Small intestine resting tone is
increased, with periodic spasms, but the amplitude of nonpropulsive
contractions is markedly decreased. In the large intestine, propulsive
peristaltic waves are diminished and tone is increased; this delays passage of
the fecal mass and allows increased absorption of water, which leads to
constipation. The large bowel actions are the basis for the use of opioids in
the management of diarrhea, and constipation is a major problem in the use of
opioids for control of severe cancer pain.
c. Biliary tract—The opioids contract
biliary smooth muscle,which can result in biliary colic. The sphincter of Oddi
may con-strict, resulting in reflux of biliary and pancreatic secretions and
elevated plasma amylase and lipase levels.
d. Renal—Renal function is
depressed by opioids. It is believedthat in humans this is chiefly due to
decreased renal plasma flow. In addition, μ opioids have been found to have an
antidiuretic effect in humans. Mechanisms may involve both the CNS and
peripheral sites. Opioids also enhance renal tubular sodium reab-sorption. The
role of opioid-induced changes in antidiuretic hor-mone (ADH) release is
controversial. Ureteral and bladder tone are increased by therapeutic doses of
the opioid analgesics. Increased sphincter tone may precipitate urinary
retention, espe-cially in postoperative patients. Occasionally, ureteral colic
caused by a renal calculus is made worse by opioid-induced increase in ureteral
tone.
e. Uterus—The opioid analgesics
may prolong labor. Themechanism for this action is unclear, but both peripheral
and central actions of the opioids can reduce uterine tone.
f. Neuroendocrine— Opioid analgesics
stimulate the release ofADH, prolactin, and somatotropin but inhibit the
release of luteinizing hormone. These effects suggest that endogenous opi-oid
peptides, through effects in the hypothalamus, regulate these systems (Table
31–1).
g. Pruritus—Therapeutic doses of
the opioid analgesics produceflushing and warming of the skin accompanied
sometimes by sweating and itching; CNS effects and peripheral histamine release
may be responsible for these reactions. Opioid-induced pruritus and
occasionally urticaria appear more frequently when opioid analgesics are
administered parenterally. In addition, when opioids such as morphine are
administered to the neuraxis by the spinal or epidural route, their usefulness
may be limited by intense pruritus over the lips and torso.
h.
Miscellaneous— The opioids modulate the immune systemby effects on lymphocyte
proliferation, antibody production, and chemotaxis. In addition, leucocytes
migrate to the site of tissue injury and release opioid peptides, which in turn
help counter inflammatory pain. However, natural killer cell cytolytic activity
and lymphocyte proliferative responses to mitogens are usually inhibited by
opioids. Although the mechanisms involved are com-plex, activation of central
opioid receptors could mediate a sig-nificant component of the changes observed
in peripheral immune function. In general, these effects are mediated by the
sympathetic nervous system in the case of acute administration and by the
hypothalamic-pituitary-adrenal system in the case of prolonged administration
of opioids.
C. Effects of Opioids with Both Agonist and Antagonist Actions
Buprenorphine is an
opioid agonist that displays high binding affinity but low intrinsic activity
at the μ
receptor. Its slow rate of dissociation from the μ receptor has also made it an attractive
alternative to methadone for the management of opioid with-drawal. It functions
as an antagonist at the δ and κ receptors and for
this reason is referred to as a “mixed agonist-antagonist.” Although
buprenorphine is used as an analgesic, it can antagonize the action of more
potent μ
agonists such as morphine. Buprenorphine also binds to ORL1, the orphanin
receptor. Whether this property also participates in opposing μ receptor function is
under study. Pentazocine and nalbuphine are other examples of opioid analgesics
with mixed agonist-antagonist prop-erties. Psychotomimetic effects, with
hallucinations, nightmares, and anxiety, have been reported after use of drugs with
mixed agonist-antagonist actions.
A combined
buprenorphine HCl/naloxone HCl dihydrate preparation is now available as
sublingual tablets and a sublingual film for use in a maintenance treatment
plan that includes counsel-ing, psychosocial support, and direction by
physicians qualified under the Drug Addiction Treatment Act. Both formulations
can be abused in a manner similar to other opioids, legal or illicit. The
combination formulations can cause serious respiratory depression and death,
particularly when extracted and injected intravenously in combination with
benzodiazepines or other CNS depressants (ie, sedatives, tranquilizers, or
alcohol). It is extremely dangerous to self-administer benzodiazepines or other
CNS depressants while taking the buprenorphine-naloxone combination.
Related Topics