Chapter: Essential Clinical Immunology: Immunological Aspects of Immunodeficiency Diseases
Phagocyte Deficiencies
PHAGOCYTE DEFICIENCIES
Neutrophils are the principal circulating phagocyte. During inflammation, neutro-phils become activated and migrate into the tissues where they ingest, kill, and digest invading bacteria and fungi. Neu-trophil function can be deficient because of a reduction in the number of circulating neutrophils (neutropenia) or due to inher-ited defects in neutrophil function, which are rare disorders.
Neutropenia
Neutropenia is defined as a decrease in the blood neutrophil count below 1.5 Ă— 109/ L. In some racial groups (e.g., Africans), the normal neutrophil count is somewhat lower (up to 1.2 Ă— 109/L). While mild neu-tropnia may be asymptomatic, severe neu-tropenia (counts < 0.5 Ă— 109/L) is invariably associated with the risk of life-threatening
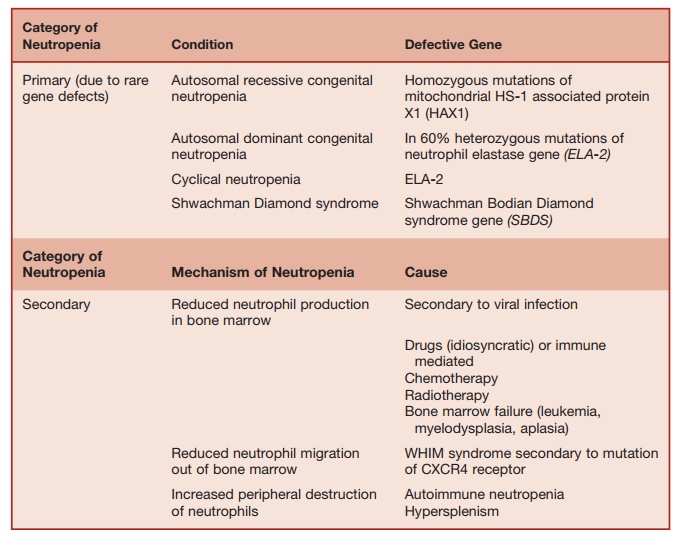
Table 5.7 Causes of Neutropenia
Defects in Leucocyte Migration
To reach the sites of inflammation, neutrophils need to migrate across the endo-thelium into sites of inflammation. For this process to be initiated, sialyl LewisX, which is expressed on the surface of leucocytes,
needs to interact with E selectin, which is expressed on the luminal surface of the endothelial cells. Leucocytes, which are transiently arrested by the previous inter-action, need to bind tightly to the endothe-lial surface by a second set of interactions. This is between the protein lymphocyte function-associated antigen-1(LFA-1) LFA-1 expressed on the leucocyte surface and its ligand intercellular adhesion molecule-1 expressed on the luminal surface of acti-vated endothelial cells. Leucocyte emigra-tion into the tissues follows these adhesion events. LFA-1 is one of a set of three cell-surface heterodimers composed of a com-mon β chain (CD18) with three separate α chains called CD11a, b, and c. CD18-CD11a heterodimers form LFA-1, CD18/CD11b
Table 5.8 Defects in Phagocyte Function
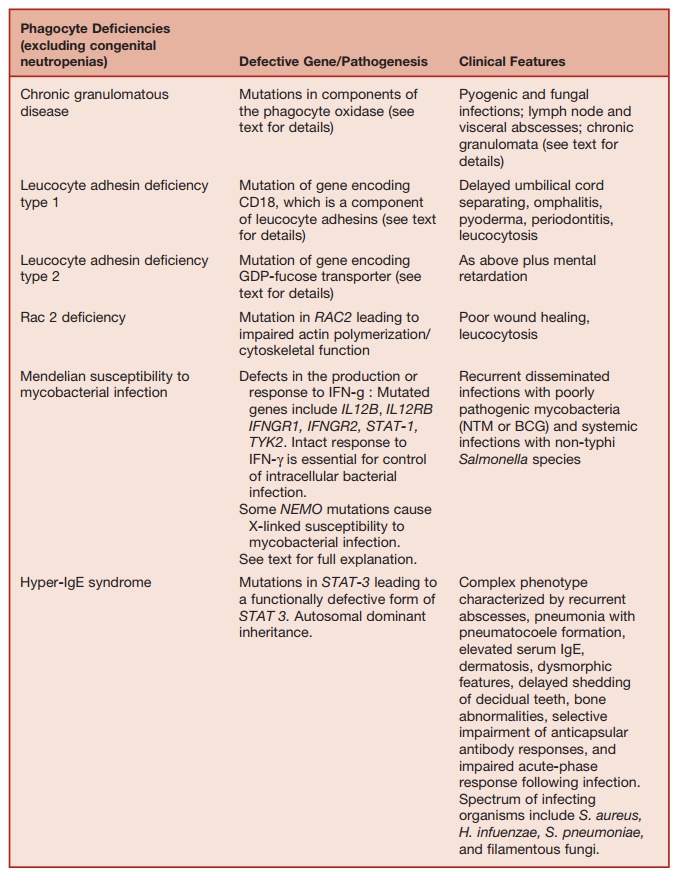
LFA-1 is required for leucocyte adhesion to endothelial cells while CR3 and CR4 act as receptors for activated complement, aiding ingestion of opsonized microorganisms. Mutation of the gene encoding CD18 (resulting in the lack of expression of LFA-1, CR3, and CR4) results in an inherited primary immunode-ficiency called leucocyte adhesin deficiency type 1(LAD1). Mutations of the enzyme GDP-fucosyl transferase prevents post-transcriptional fucosylation of proteins. Such individuals cannot synthesize sialyl LewisX. This condition is called leucocyte adhesion deficiency type 2. Leucocytes of the patient with LAD1 and 2 exhibit impaired ability to adhere to endothelial walls and therefore cannot migrate into infected sites.
These patients typically present in early childhood with recurrent pyogenic infection of skin, respiratory, and gastro-intestinal tracts as well as mucous mem-branes. Poor wound healing and delayed umbilical cord separation are typical. Because of impaired neutrophil migration, these patients develop a leucocytosis and pus fails to form at sites of infection. These inherited disorders are typically associated with severe gingivitis and periodontal disease, again indicating the particular importance of normal neutrophil function for maintenance of the health of the den-tal crevice. Outcome in both conditions is poor, with early death. BMT is curative in LAD1, and oral fucose supplementation is beneficial in LAD2.
Defects in Bacterial Killing
The best example of an immunodeficiency characterized by failure of phagocyte-mediated bacterial killing is chronic granulomatous disease (CGD). In normal neutrophils (and monocytes), bacterial phagocytosis results in the activation of the nicotinamide adenine dinucleotide phos-phate (NADPH) oxidase complex, which produces superoxide and generates a mi-lieu within the phagosome that activate
the proteolytic enzymes, cathepsin, and elastase. The activity of the proteolytic enzymes is bactericidal. The NADPH oxi-dase complex comprises two membrane-associated proteins, p91phox and p22phox (also called the α and β units of cytochrome B558, respectively), complexed with three cytosolic cofactors, p47phox, p40phox, and p67phox. Mutations have been identified in four out of five components (p91phox, p22phox, p47phox, p67phox), resulting in defective NADPH oxidase activity, leading to impairment of bacterial kill-ing by phagocytic cells. The result is a clinical syndrome, called CGD, character-ized by failure of bacterial degradation in vivo, resulting in the persistence of tissue inflammation with granuloma formation in a variety of organs. CGD due to p91phox deficiency is X-linked, while the other vari-ants of CGD are inherited in an autosomal recessive manner.
In CGD, the bacterial killing mecha-nisms that depend on the phagocyte oxidase system are inoperative, but the nonoxidative killing mechanisms are still intact. Therefore, patients with CGD are not troubled by the broad range of microbes that a neutropenic patient would be susceptible to. Instead, CGD patients get infections with a restricted range of micro-organisms, which are only susceptible to the bactericidal mechanisms initiated by NADPH oxidase activation. The spectrum of microorganisms that causes infections in CGD include S. aureus, Gram-negative bacteria (Burkholderia cepacea, Salmonella, Serratia), and fungi (Aspergillus spp.). Characteristic sites of infection include subcutaneous tissue, lymph nodes, lungs, and liver. Oral and perioral ulceration are common.
The formation of chronic granulomata in various tissues is a typical feature of CGD. In critical locations, chronic granu-loma formation may cause pathology. Granulomatous obstruction of the gastro-intestinal or genitourinary tract may be a consequence. Hepatosplenomegaly may occur due to granulomatous infiltration of these organs. A granulomatous colitis, resembling Crohn’s disease, occurs in about 15 percent of CGD patients, highlighting the importance of normal phagocyte func-tion for preventing harmful inflammation in the large intestine.
Diagnosis of CGD is based on the abil-ity of stimulated neutrophils from these patients to oxidize dyes to give a colored product or a fluorescent product, detect-able by flow cytometry. Prophylactic treat-ment with IFN-Îł substantially reduces the incidence of bacterial and fungal infections in CGD patients, for reasons that are not fully understood.
The cytosolic GTPase Rac2 is required for the normal actin polymerization and optimal function of the phagocyte oxidase system. Rac2 deficiency results in impaired neutrophil mobility and poor superoxide responses to some stimuli. A single affected patient identified to date had a clinical presentation similar to that seen in LAD.
Defects in Killing of Intracellular Bacteria by Activated Macrophages
Some bacterial species (Mycobacteria, Lis-teria, Salmonella) are resistant to the killing mechanisms operating within phagocytic cells and therefore can survive and multi-ply within monocytes and macrophages. Effective immunity against these organ-isms depends on T-cell- (and NK-cell-) dependent macrophage activation. Studies in gene-disrupted mouse and human immunodeficiencies have identified that
IL-12- and IL-23-dependent interferon gamma (IFN-γ) production is critically important for immunity against intracel-lular bacterial pathogens. This process is initiated by the stimulation of Toll recep-tors on the surface of antigen-presenting cells by bacterial ligands such as mycobac-terial lipoarabinomannan. This results in the secretion of IL-12 and IL-23 and TNF-α by the antigen-presenting cells. Binding of IL-12 and IL-23 to their respective recep-tors, expressed on the surface of activated T cells and NK cells, induces these cells to secrete a further cytokine IFN-γ. IFN-γ acting in concert with TNF-α activates macrophages, which are then capable of killing intracellular pathogens (e.g., Myco-bacteria and Salmonella). Individuals with impaired T-cell-mediated immunity (e.g., SCID or HIV infection) are highly suscep-tible to mycobacterial infections, including infections caused by poorly pathogenic mycobacterial species (nontuberculous mycobacteria and BCG). However, dissem-inated life-threatening infections by these organisms may also occur in the absence of a recognized primary or secondary immu-nodeficiency. Genetic analysis of affected kindred to date have identified mutations in seven different genes that participate in the production or response to IFN-γ. These patients are classified under the heading Mendelian susceptibility to mycobacte-rial disease (MSMD) (OMIM 209950). The genetic lesions responsible for MSMD affect the integrity of the IL-12/23-depen-dent IFN-γ pathway.
Thus, mutations have been identified in
â– The P40 subunit shared by IL-12 and IL-23 (also called IL12B);
■The β chain shared by the IL-12 and IL-23 receptors;
â– The TYK2 kinase required for signaling via the IL-12 receptor;
■The α and β chains of the IFN-γ receptor (IFN-γ R1 and IFN-γ 2);
â– The STAT-1 signal transducing molecule, which is required for signaling through the IFN-Îł receptor;
■The seventh defect responsible for increased susceptibility to mycobacterial infection is that NFκB essential modulator gene, which is required for NFκB activation, which is critical for signal transduction via the Toll, IL-1, and TNF-α receptors.
The severity of the clinical phenotype of MSMD depends on the genotype. Com-plete IFN-ÎłR1 or R2 deficiencies lead to the abrogation of responses to IFN-Îł. Such patients present in early childhood with disseminated NTM or BCG infections, resulting in a high mortality, despite chemotherapy. Mycobacterial lesions in such patients are multibacillary and asso-ciated with poor granuloma formation. In contrast, partial IFN-Îł R1 deficiency, complete IL-12B deficiency, and IL-12/23 receptor deficiency predispose to myco-bacterial infections, presenting at a later age and with a favorable outcome, fol-lowing chemotherapy. Lesions in these latter patients are paucibacillary and are associated with an intact granulomatous response. Patients with partial or com-plete STAT-1 deficiency, which impairs the signaling via the IFN-Îł R results in susceptibility to mycobacteria of low-grade virulence. Complete STAT-1 defi-ciency also impairs signaling via type I interferon receptors, which predisposes to life-threatening herpes viral infections occurring in childhood. The commonestinfections that affect patients with defects in the IL-12/23 are extraintestinal or sep-ticemic, relapsing infections with non-typhoidal Salmonella species.
As could be predicted, patients with IL-12/23 defects or partial IFN-ÎłR defects respond to treatment with IFN-Îł but those with complete IFN-Îł defects are refractory to this therapy.
Related Topics