Chapter: Essential Clinical Immunology: Immunological Aspects of Immunodeficiency Diseases
Immunodeficiencies Resulting in Defective Homeostasis of the Immune System
IMMUNODEFICIENCIES RESULTING IN DEFECTIVE HOMEOSTASIS OF THE IMMUNE SYSTEM
Defects in the Cytolytic Pathway
The homeostasis of immune responses requires prevention of excessive lymphocyte activation (Table 5.9). The mecha-nism by which such regulation occurs includes activation-induced cell death of T lymphocytes, which requires the activa- tion of apoptotic pathways. Defects in criti-cal components of these pathways result in susceptibility to hemophagocytic lym-phohistiocytosis (HLH), which is usually triggered by an intercurrent viral infection caused by viruses such as EBV or cytomeg-alovirus. HLH is characterized by massive infiltration of organs, such as the liver, spleen, bone marrow, and central nervous system, by activated CD8+ lymphocytes and macrophages, as well as a massive overproduction of IFN-╬│ and TNF-╬▒. Severe pancytopenia is typical of this syndrome and is caused in part by phagocytosis of blood cells by activated macrophages and in part is secondary to the infiltration of bone marrow by activated macrophages (histiocytes).
A number of genetic defects that affect the efficiency of T-cell- and NK-cell-medi-ated cytolysis can predispose to the devel-opment of HLH. Cytolysis by T cells and NK cells is initiated by the secretion of contents of cytolytic granules at the immu-nological synapse between the T cells and the target cells. The process involves the translocation of perforin-containing lytic granules onto the target cell inter-phase, followed by fusion of these gran-ules with the plasma membrane of the T cell, with release of perforin onto the sur-face of the target cell. Perforin punches holes in the target cell membrane, causing cytolosis.
Mutation of the perforin (PRF1) gene, which encodes for perforin, is one of the genetic defects that predisposes to famil-ial HLH. Perforin-supported cytolysis (by CD-8 cells and NK cells) may damp down immune responses triggered by viral infections by aiding the elimination of antigen-presenting cells or by promoting activation-induced death of T cells.
Table 5.9 Disorders of Homeostasis of Immune Function
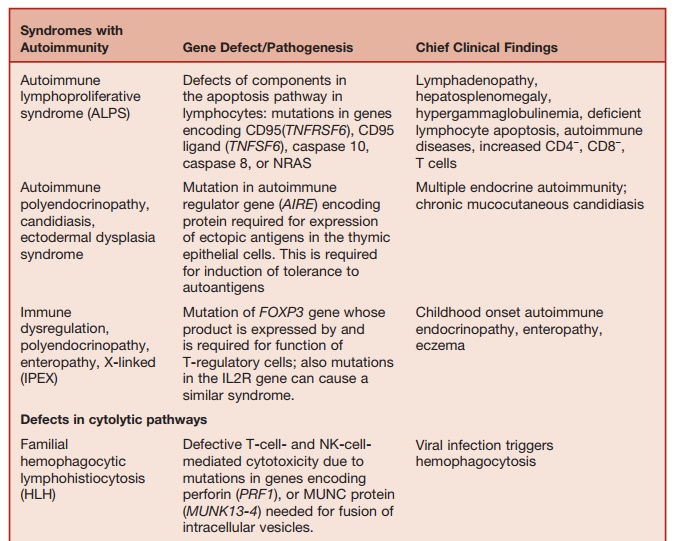
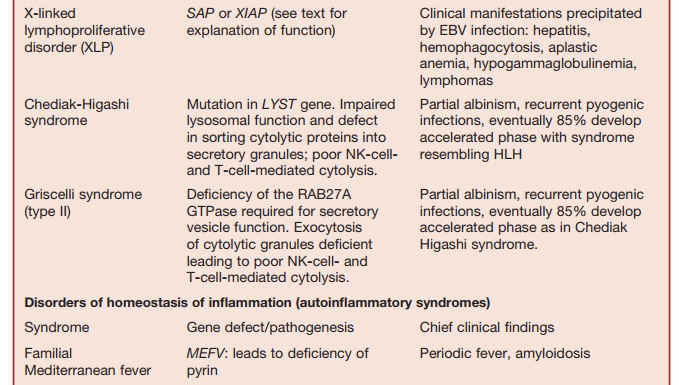
Studies of rare immunodeficiency syn-dromes, all of which are characterized by an increased tendency to develop HLH have identified a number of components required for the normal expression of cytolytic capacity by T cells and NK cells. The intracellular migration and docking of lytic granules requires the function of the small Rab GTPase, RAB27, which is mutated in the Griscelli syndrome char-acterized by partial albinism, immunode-ficiency, and susceptibility to developing HLH. Defective cytolytic granule exocy-tosis is also characteristic of patients with mutations of the gene encoding the protein MUNK13-4 (UNC13D). The tSNARE syn-texin 11, which is present in the trans-Golgi network, is also involved in intracellular vesicle trafficking. Mutation of these two genes is responsible for a further form of familial HLH.
Lysosomal trafficking regulator is defi-cient in individuals with a mutation in the CHS1 gene. This causes a defect in sorting of cytolytic proteins into secretory granules. This is the underlying defect in the condition called Chediak-Higashi syndrome, which is another condition characterized by susceptibility to HLH.
Immunodeficiencies Characterized by Increased Susceptibility to EBV Infections
XLP is a rare inherited immunodeficiency characterized by life-threatening patho-logical processes triggered by EBV infec-tions. The manifestations of this condition include fulminant infectious mononu-cleosis, virally induced HLH, hypogam-maglobulinemia, and the propensity to develop malignant lymphomas. Mutation in the gene SH2D1A encoding the signaling lymphocyte activation molecule (SLAM) associated protein (SAP) are responsible for more than 60 percent of cases of XLP. SH2D1A ŌĆō/ŌĆō mice lack NK T cells and this defect could be corrected by reconstitution
of SH2D1A expression within bone mar-row cells derived from SH2D1A ŌłÆ/ŌłÆ mice. Patients with XLP also lack NK T cells. Therefore, it is possible that NK T-cell function is essential for controlling EBV infection as well as homeostasis of T-cell responses to this virus. Further support for this theory comes from the recent observation that XLP can also be caused by a mutation in the gene (BIRC4) that encodes for the protein X-linked inhibitor of apoptosis (XIAP). XIAPŌłÆ/ŌłÆ patients also have reduced numbers of NK T cells.
The pathological manifestations of the XLP cannot be completely attributed to the reduced NK- and T-cell function seen in this condition. The SAP, which acts as a signaling adaptor molecule within lymphoid cells, and XIAP, which regulates apoptosis, help in the homeo-stasis of immune responses in complex ways. Therefore, mechanisms underlying the pathogenesis of XLP are likely to be complicated.
Immunodeficiencies Characterized by Increased Liability to Develop Autoimmunity
Immune responses to self-antigens are pre-vented by:
1. The elimination of self-reactive B and T cells in the thymus and bone marrow (central tolerance).
2. Inhibition of self-reactive T and B cells that escape central tolerance induc-tion, by the action of regulatory T cells found in the periphery. These T-regulatory cells, which are CD25+ and CD4+, are required for peripheral tol-erance and the active suppression of autoimmunity.
IMMUNE DYSREGULATION POLYENDOCRINOPATHY AND ENTEROPATHY, X-LINKED SYNDROME
The product of the FOXP3 gene (belong-ing to the forkhead family) is required for the generation and the function of CD4+ CD25+ T-regulatory cells. Mice that are FOXP3 ŌłÆ/ŌłÆ lack these cells and develop a syndrome characterized by lymphoprolif-eration and autoimmunity. Expression of ectopicFOXP3 confers suppressive func-tion on CD4+ and CD25+ T cells. Human males with mutations of the FOXP3 gene (which is located on the X chromo-some) develop a syndrome characterized by neonatal endocrinopathies (including type I diabetes) enteropathy, eczema, immune thrombocytopenia, and cachexia. In these patients, pancreatic islet cell and intestinal mucosal damage are secondary to infiltration of the tissues with mononu-clear cells and plasma cells, signifying an autoimmune pathogenesis. This condition is called the immune dysregulation poly-endocrinopathy and enteropathy, X-linked (IPEX) syndrome. This condition provides evidence supporting current concepts on the role of T-regulatory cells in preventing autoimmunity.
Recently, mutations in the IL2R gene encoding for the ╬▒ chain of the IL-2 receptor have been described in patients with the clinical phenotype of the IPEX syndrome. CD4 T cells of these patients fail to produce the immunosuppressive cytokine IL-10.
AUTOIMMUNE LYMPHOPROLIFERATIVE SYNDROME
During lymphocyte development, autoreactive cells are culled by undergo-ing apoptotic death. During the life span of mature lymphocytes, activation and effector function is followed by activation- induced cell death by apoptosis. Apop-tosis thus maintains homeostasis in the immune system by minimizing autoim-mune reactions to self-antigens, as well as limiting the total size of the peripheral lymphocyte pool. Antigen-mediated acti-vation of T cells induces them to express a surface receptor FAS (CD95) as well as its ligand, the FAS-ligand. FAS/FAS-ligand interaction activates a biochemi-cal pathway culminating in cell death by apoptosis. Mice that are FAS ŌłÆ/ŌłÆ (lpr) or FAS-ligand ŌłÆ/ŌłÆ (gld) develop autoimmu-nity and develop expansion of their their lymphocyte pool.
ALPS is a rare disease caused by defective cellular apoptosis. Failure of lymphocytes to undergo apoptosis results in impaired homeostasis of the lymphoid population. Patients develop lymphocytosis, hyperplasia of lymphoid organs (spleen, lymph nodes), hyper-gammaglobulinemia, and autoimmunity (autoimmune cytopenias, Guillain-Barr├® syndrome). Peripheral blood and periph-eral lymphoid tissues characteristically╦£
Mutations in the FAS-mediated pathway of apoptosis are responsible for most cases. These include mutations of FAS, FAS-ligand, and cas-pase 10. Caspase 8 deficiency causes impaired CD95-mediated apoptosis as well as a defect in T-, B-, and NK-cell function, manifesting as an immunode-ficiency. Recently, an activating muta-tion in the NRAS gene encoding a GTP-binding protein with a broad spectrum of signaling functions has been found in other patients with ALPS. Defective, IL-2 withdrawal induced, apoptosis of lym-phocytes is a characteristic abnormality in these patients.
Autoinflammatory Syndromes
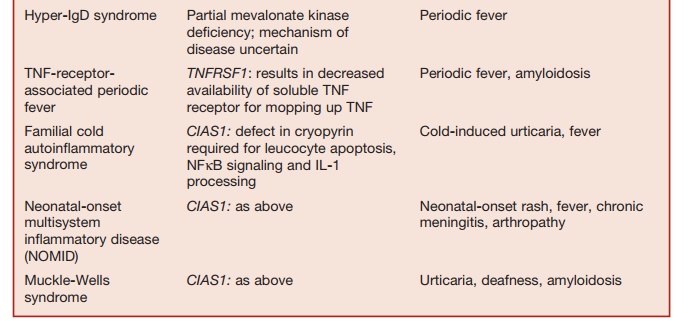
The responses of acute inflammation and fever are protective responses triggered by infection or tissue damage, acting through the innate immune system. The study of a collection of disorders termed familial periodic fever syndromes has contributed to our understanding of the physiological processes underlying the homeostasis of inflammation and fever. These inherited disorders are characterized by recurrent episodes of fever and systemic inflamma-tion. These conditions are not associated with autoantibody production or self-reactive T-cell responses. They are col-lectively called autoinflammatory syndromes. Unraveling the pathophysiology of these disorders has given rise to the concept that these conditions are due to an innate immune system that is either oversensitive and prone to activation by minor stimuli or is poorly regulated.
The cleavage of pro-IL-1b to its active product IL-1 by the action of caspase-1 (interleukin converting-enzyme) is a key event in the generation of an acute inflam-matory response. Activated caspase-1, intern, is generated from pro-caspase-1 by the action of activated forms of two cyto-solic proteins called pyrin and cryopyrin, acting via adaptor proteins. The best-char-acterized adaptor protein is called ASC (apoptosis-associated speclike protein with a caspase recruitment domain). As summarized in Table 5.9, mutations of components of this system (also called an inflammasome) give rise to most auto-inflammatory syndromes characterized to date. The net effect of all these muta-tions is to impair homoeostasis of the pro-inflammatory cytokine IL-1, NF╬║B acti-vation, and cellular apoptosis. Impaired cellular apoptosis leads to the persistence of activated leucocytes that would other-wise undergo apoptosis, leading to the resolution of inflammation. Collectively, these abnormalities permit the inappro-priate amplification and persistence of inflammatory responses leading to the clinical features of the autoinflammatory syndromes.
Related Topics