Chapter: Essential Clinical Immunology: Immunological Aspects of Immunodeficiency Diseases
Major Categories of Antibody Deficiency
Major Categories of Antibody Deficiency
These are summarized in Table 5.4.
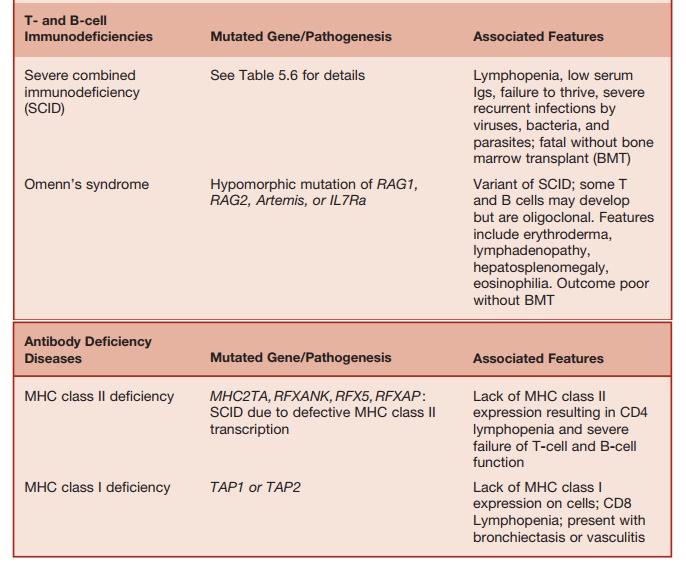
Table 5.4 Major Antibody and/or T-cell Deficiencies
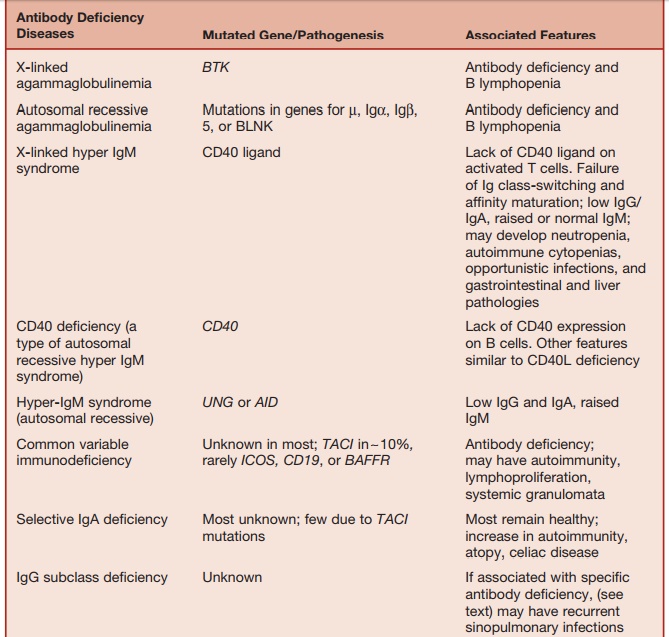
ANTIBODY DEFICIENCY ASSOCIATED WITH ABSENT B CELLS
B-cell maturation beyond the pre-B-cell stage found in the bone marrow, requires signals received through the pre-B-cell receptor complex. The pre-B-cell receptor is composed of the ┬Ą chains, surrogate light chains (heterodimers of ╬╗ constant region
The activities of the protein BTK (BrutonŌĆÖs tyrosine kinase) and BLNK (B-cell linker protein) are also essential for the transduction of signals received via the B-cell (and pre-B-cell) receptors. Therefore, it is not surprising that mutations in each of these elements causes early-onset antibody deficiency associated with lack of circulating B cells.
Ninety percent of all such cases occur in boys due to mutation of the BTK gene, which maps to the X chromosome (Xp22). This condition is called X-linked agammaglobulinemia, which was the first immunodeficiency to be described in 1952 by Colonel Ogden Bruton. Mutations in the genes, encoding ╬╝, 5, Ig╬▒, Ig╬▓, and BLNK cause rare autosomal recessive forms of early-onset antibody deficiency with severe B lymphopenia.
ANTIBODY DEFICIENCY DUE TO DEFECT IN IMMUNOGLOBULIN ISOTOPE SWITCHING
During primary antibody responses, B cells initially produce IgM and later on in the response switch to the production of IgG, IgA, and IgE.
During the so-called immunoglobulin class-switching process, the heavy chain constant region changes while antigen specificity is maintained. Immunoglobulin class switch takes place within germinal centers contained within B-cell follicles of the secondary lymphoid organs. Another process that occurs within germinal centers is somatic hypermutation, which results in the sequential accumulation of point muta-tions in the Ig variable region gene. If the point mutation(s) result in increased bind-ing affinity to the inducing antigen, the B-cell blasts (centrocytes) survive, proliferate, and eventually give rise to memory B cells and plasma cells that secrete high-affinity antibody (this process is called affinity maturation). Through these processes, memory B cells are generated within germina centers.
Defects in genes encoding molecules required for the above processes, which operate within germinal centers, result in a form of antibody deficiency with elevated (or normal) IgM levels but lack-ing IgG, IgA, and IgE. These conditions are called hyper-IgM (HIGM) syndromes. A key requirement for germinal center formation and function is the interaction of CD40 (belonging to the TNF-receptor superfamily) found on the surface of B cells with an ŌĆ£activation induced,ŌĆØ CD40-ligand (CD40L) protein expressed on the surface of CD4 lymphocytes. Mutations in the CD40L gene or the CD40 gene result in X-linked (relatively common) and autoso-mal recessive (rare) HIGM, respectively. Patients with the CD40L deficiency suffer from recurrent bacterial infections typical of antibody deficiency. However, because CD40L function is required for optimal T-cell immunity, they also suffer from opportunistic infections characteristic of T-cell deficiency. About a third of patients with CD40L deficiency develop Pneumocys-tis pneumonia. Infections with cryptosporio-dosis, toxoplasmosis, and nontuberculous
mycobacteria also occur in this condition. These opportunistic infections can be ex-plained on the basis that the interaction of CD40L on activated T cells with CD40 ex-pressed on the surface of macrophages and dendritic cells, which in turn undergo matu-ration and activation, is required for the opti-mum expression of antimicrobial immunity.
A high proportion of CD40L-deficient patients develop progressive liver dam-age (sclerosing cholangitis), probably the result of cryptosporidial infection of the bile ducts.
Defects in the RNA-editing enzymes, activation-induced cytidine deaminase (AID), and uracil-DNA glycosylase (UNG) result in two further types of HIGM syn-dromes, which are defective class switch-ing and affinity maturation.
Signaling through CD40, which belongs to the TNF-receptor superfamily, depends on the activation of the inhibitor of ╬║ kinase (IKK) complex, resulting in the induction of NF╬║B. Hypomorphic mutations of the gamma subunit of the IKK complex, which is called NEMO (NF╬║B essential modu-lator), impairs NF╬║B activation. Patients with mutations in NEMO develop a com-plex immunodeficiency, which includes features of the HIGM syndrome.
COMMON VARIABLE IMMUNE DEFICIENCY
Most patients with primary antibody deficiency are collected under the heading ŌĆ£common variable immune deficiency,ŌĆØ which is a condition characterized by low serum IgG and IgA and a variable decrease in IgM and the impaired production of specific antibodies following natural microbial exposure or immunization. The estimated prevalence of CVID is between 1 in 10,000 and 1 in 50,000. Clinically, CVID is dominated by the effects of antibody deficiency. About 20 percent of patients with CVID develop autoimmune disor-ders (arthritis, cytopenias, endocrinopa-thies). Some patients with CVID develop noncaseating sarcoidlike granulomata infiltrating various organs (lungs, liver, spleen, skin). The mechanisms underlying autoimmunity and granulomatous disease in CVID are unknown.
The immunological phenotype of CVID is heterogeneous with documented defects in B-cell survival, generation of B memory cells, and in vitro B- and T-cell activation. About 10 percent of cases of CVID are familial, with a predominance of autosomal dominant or autosomal reces-sive inheritance. CVID or selective IgA deficiency can affect different members of the same kindred. Recently, a number of gene defects have been identified in CVID patients, accounting for about 10ŌĆō15 per-cent of the total patient pool.
The commonest defect (in approxi-mately 10% of CVID patients) is a mutation of the gene TACI (transmembrane activator and calcium modulator and cyclophilin ligand interacter). TACI, which belongs to the TNF-receptor superfamily, is a ligand for the cytokines, BAFF (B-cell-activat-ing factor of the TNF family) and APRIL (a proliferation-induced ligand), which induce immunoglobulin class-switch recombination.
Mice deficient in BAFF or its recep-tor have impaired B-cell development and antibody deficiency. TACIŌłÆ/ŌłÆ mice have reduced serum IgA and IgM levels, reduced antibody responses to T-cell-independent antigens, and tend to develop autoimmunity and B lymphoprolifera-tion. Mutations in TACI have been found in CVID patients and their relatives, with selective IgA deficiency, indicating variable penetrance of this gene defect. In the majority of currently documented patients, TACI mutations affect only one allele, indicating a dominant negative effect of the mutated gene. A possible explana-tion is that TACI, like other members of the TNF-receptor family, undergoes ligand-independent preassociation and function as multimeric units. Thus, incorporation of a mutated TACI chain in this multimeric complex may disrupt ligand binding or signal-transducing capacity. Recent stud-ies have found that family members of CVID patients possessing heterozygous TACI mutations may have completely nor-mal serum immunoglobulin levels and ŌĆ£in vitroŌĆØ B-cell function. This leads to the con-clusion that the development of antibody deficiency in those carrying heterozygous TACI mutations may depend on modifier genes or environmental factors.
ICOS (inducible co-stimulating recep-tor) is a co-stimulatory T-cell molecule that induces cytokines required for sup-porting class-switch recombination, Ig production and terminal B-cell differentia-tion. ICOSŌłÆ/ŌłÆ and ICOS ligandŌłÆ/ŌłÆ mice exhibit defective germinal center formation and antibody production. Mutations in the ICOS gene account for about 1 percent of CVID patients.
A single CVID patient was identified to have a mutation on the gene encoding the BAFF receptor, a finding that could be pre-dicted from observations in BAFFŌłÆ/ŌłÆ mice.
Finally, CD19 deficiency has been described in two families with CVID. CD19 is a B-cell accessory molecule that is required for B-cell activation, proliferation, and hence B-cell development. CD19ŌłÆ/ŌłÆ mice exhibit hypogammaglobulinemia and poor antibody responses to protein antigens.
In the case of the large majority of patients with CVID, the underlying molec-ular defect is unknown.
IgA DEFICIENCY
IgA deficiency is characterized by reduced (<0.07 g/L) or absent serum IgA levels. IgA deficiency is the commonest form of primary antibody deficiency and affects about 1 in 700 Caucasians. IgA deficiency is rare in some racial groups (Japanese, Africans). The majority of IgA-deficient individuals remain free of infec-tion due to the ability of IgG and IgM to compensate for the lack of IgA. Long-term studies have shown that a small propor-tion of IgA-deficient patients develop recurrent sinopulmonary or gastroin-testinal infections. Most infection-prone IgA-deficient patients have concomitant IgG2 subclass deficiency and a selective inability to produce antibodies against bacterial capsular polysaccharides. IgA deficiency is associated with an increased incidence of atopy, celiac disease, and a range of autoimmune diseases, including arthritis, ŌĆ£lupus-likeŌĆØ syndrome, autoim-mune endocrinopathies, and autoimmune cytopenias.
IgA-deficient patients with serum lev-els below 0.07 g/L are at the risk of devel-oping anti-IgA antibodies after receiving blood products. Such individuals are at risk of developing anaphylaxis on receiv-ing blood products.
IgA deficiency and CVID can differ-entially affect members of the same kin-dred. Rarely, IgA deficiency can precede the development of CVID. Therefore, in some instances, the molecular mechanisms underlying CVID and IgA deficiency may be identical. TACI deficiency can cause IgA deficiency in some family members while others develop CVID. However, the molec-ular basis of IgA deficiency in most cases is largely unknown.
Genetic analysis of kindred with CVID and IgA deficiency have highlighted the existence of susceptibility loci within the MHC region of chromosome 6. The stron-gest linkage lies within the DR/DQ within the MHC class II region.
IgG SUBCLASS DEFICIENCY
Serum IgG comprises four subclasses called IgG1, IgG2, IgG3, and IgG4, respec-tively reflecting the relative abundance of these isotypes in the serum. IgG sub-class deficiency is diagnosed on finding a reduction in the serum concentration of IgG subclasses, of more than two standard deviations below the mean value for age, despite the total IgG level being normal. In practice, IgG subclass assays are difficult to standardize due to the lack of an inter-nationally accepted reference preparation. Furthermore, genetic variations that influ-ence serum IgG subclass levels exist among different ethnic groups. Hence, age- and population-related normal ranges are not always available.
Some individuals with IgG subclass deficiencies are asymptomatic. Others with IgG subclass deficiencies are prone to recurrent sinopulmonary infections. Such infection-prone patients exhibit reduced antibody responses to bacterial capsular polysaccharides. Defective anti-polysac-charide antibody responses are most often seen in individuals with IgG2 subclass deficiency with or without concomitant IgA deficiency. The molecular mechanisms underlying IgG subclass deficiencies are unknown.
TREATMENT OF ANTIBODY DEFICIENCY
Prospective clinical studies have shown that optimal IgG replacement ther-apy reduces the incidence of sepsis and complications like structural lung dam-age. If replacement therapy is introduced early before organ damage is established,
antibody-deficient individuals are likely to have a normal life span. Replacement immunoglobulin is prepared from plasma pooled from large cohorts of normal donors. Current practice of prescreening donors for infection and multiple antivi-ral steps employed during the manufac-ture of plasma have eliminated the risk of HIV and hepatitis B and C transmission. Different immunoglobulin products are licensed for administration via the intra-venous or subcutaneous route. Detailed management of a patientŌĆÖs antibody defi-ciency is discussed in the references on the diagnosis and management of anti-body deficiency.
Related Topics