Chapter: Essential Clinical Immunology: Immunological Aspects of Immunodeficiency Diseases
Inherited Deficiency of the Complement System
INHERITED DEFICIENCY OF THE COMPLEMENT SYSTEM
Activation of the complement system underlies one of the main effector pathways contributing to antibody-mediated immunity. The normal function of the comple-ment system includes defending the body against pyogenic bacterial infection and helping to eliminate immune complexes and damaged cells (Table 5.10).
Table 5.10 Complement Deficiency
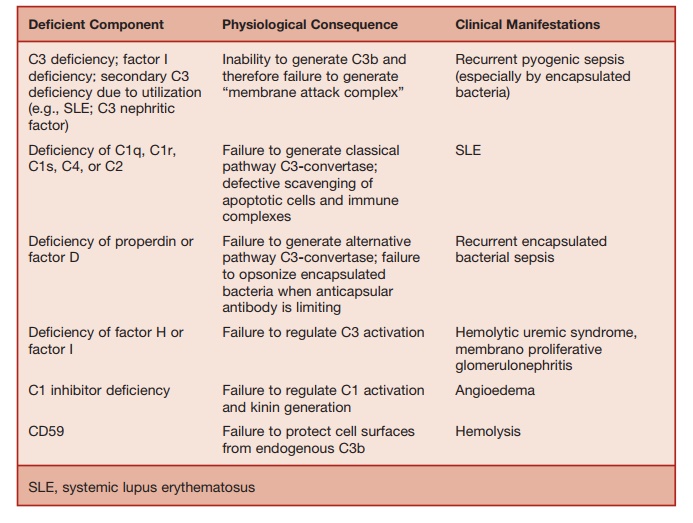
The key event in complement activation is the proteolytic cleavage of C3 to C3a and C3b. Three pathways can lead to C3 cleavage, namely, classical, alternative, and mannose-binding lectin (MBL) pathways. C3 cleavage leads on to the activation of the terminal complement pathway, caus-ing the generation of the membrane attack complex (MAC), which assembles a lipo-philic complex capable of lysing plasma membranes of susceptible cells.
The inability to generate sufficient CD3b results in increased susceptibility to pyogenic sepsis, especially infections caused by encapsulated bacteria. C3 deficiency may be due to complement utilization (e.g., in systemic lupus erythematosus [SLE] or the presence of a C3 nephritic fac-tor) or rarely autosomal recessive C3 defi-ciency. Factor I deficiency also leads to pro-found CD3 deficiency due to uncontrolled progression of C3 cleavage. Hereditary C2 deficiency and less commonly inherited C4
Protection from Neisserial infection requires the ability to generate the MAC, which lyses these bacteria. Patients with inherited homozygous deficiency of C5, C6, C7, C8, and C9 are susceptible to recur-rent meningococcal infections. In Japanese populations, an inherited terminal comple-ment component deficiency leads to about a 5,000-fold increase in the risk of Neisse-rial sepsis. Primary or secondary CD3 defi-ciency, which in turn reduces the ability to generate MAC, also results in increased susceptibility to meningococcal infection. Complement deficiency is not a risk factor in sporadic cases of meningococcal disease seen in the population at large.
MBL is a serum collectin that can bind to mannose residues on microbial cell walls. Upon binding of MBL to pathogens, two serine proteases found in serum (MBL-lectin associated serine proteases 1 and 2) become activated. These serine proteases in turn activate the classical pathway at C2 and C4. MBL deficiency may arise because of one of three point mutations in the gene encoding for this protein. Polymorphisms in the promoter region of the gene also influence serum levels of MBL. Low serum levels of MBL may result in an increased incidence of pyogenic infections in young children below two years of age. MBL defi-ciency is also overrepresented in patients with pneumococcal or meningococcal sepsis. However, the clinical significance of this finding has been disputed. This is because large, long-term prospective stud-ies of adults in Scandinavia have failed to identify increased morbidity due to infec-tious diseases in MBL-deficient individuals. Therefore, except in young children who have not yet acquired a wide repertoire of protective antibodies, MBL deficiency on
Complement Deficiency and Autoimmunity
Under physiological conditions, activation of the classical complement pathway helps in the clearance of the circulating immune complexes by the resident macrophages of the reticuloendothelial system. The surface of apoptotic cells activates the classical complement pathway, leading to their effi-cient clearance by phagocytic cells express-ing complement receptors, preventing the generation of autoimmune responses to cellular components.
Deficiency of components required for generating the classical partly C3-con-vertase may result in impairment of the
process described earlier for eliminating immune complexes and “safe” disposal of apoptotic cells. This would explain the increased incidence of SLE-like disorders in patients with inherited C1q, C1r, C1s, C4, C2, C3, factor I, or factor H deficiency.
Factor H Deficiency
Complete or partial factor H deficiency is associated with the occurrence of the hemolytic-uremic syndrome, although the precise underlying pathogenic mecha-nisms are unknown.
Cell-Surface-Based Inactivators of Complement
CD59 and CD55 are cell-surface molecules anchored by glycosylphosphatidylinositol
These two proteins inactivate any C3 con-vertase molecules deposited on cell sur-faces. Somatic mutation of the enzyme (PIGA: phosphatidylinositol glycan class A), needed to generate phosphatidyl-inositol anchors for cell-surface proteins, including CD55 and CD59, in erythroid precursors, results in a condition called paroxysmal nocturnal hemoglobinuria, which is due to the increased susceptibil-ity of red cells to complement-mediated hemolysis. Isolated CD59 deficiency also results in hemolytic anemia.
C1 Inhibitor Deficiency
C1 inhibitor is a serine-protease inhibitor that inactivates the serine esterases gener-ated by complement activation (C1r and C1s), kallikrein of the kinin system, and activated factors XI and XII of the clotting cascade. In the absence of C1 inhibitor, C1 activation results in the depletion of the serum C4 level. (This is useful from a diagnostic point of view.) C1-inhibitor deficiency also results in the inability to inactivate bradykinin, resulting from the unregulated activity of kallikrein. Produc-tion of bradykinin in the tissues results in increased vascular permeability, manifest-ing as attacks of angioedema. Angioedema of the respiratory tract can lead to death from asphyxia. Angioedema of the intes-tinal tissues results in recurrent episodes of severe abdominal pain due to partial intestinal obstruction, which can mimic an acute abdominal emergency.
C1-inhibitor deficiency arises from a het-erozygous mutation of C1INH gene, which acts in an autosomal dominant manner. The single normal gene cannot maintain the synthesis of physiologically sufficient quantities of C1 inhibitor. In 85 percent of C1-inhibitor-deficient individuals, the mutation prevents transcription of the defective gene. In 15 percent of affected individuals, the gene mutation abolishes the activity of the secreted protein. Rarely, autoantibodies to C1 inhibitor can lead to acquired C1-inhibitor deficiency.
Related Topics