Chapter: Essential Clinical Immunology: Immunological Aspects of Immunodeficiency Diseases
Defects in Cell-Mediated Immunity (T-Cell Dependent Immunity)
DEFECTS IN CELL-MEDIATED IMMUNITY (T-CELL DEPENDENT IMMUNITY)
These may be inherited (primary) condi-tions, which are rare or secondary to other pathologies (Table 5.5). HIV infection, which is an important cause of secondarily impaired T-cell immunity is discussed.
Table 5.5 Causes of T-Cell Deficiency
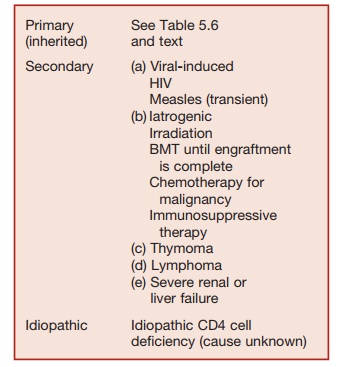
Manifestations of T-Cell Deficiency
KEY CONCEPTS
Patients with impaired T-cell function may develop the following:
1. They show increased susceptibility to infections with intracellular microbial pathogens (viruses, intracellular bacte-ria, and protozoa).
2. Viral infections: Infections with exan-thematous viruses (measles, chicken pox) can be fatal in children with T-cell deficiency. These viruses are not a problem in adults as they have residual protective antibody responses generated by primary infection or immunization. Adults with T-cell deficiency are typically affected by the reactivation of latent viruses (e.g., cyto-megalovirus, Herpes simplex), which can produce life-threatening disseminated infections.
3. Fungal infections: T-cell-deficient pa-tients are typically susceptible to fungal infections.
a. Pneumocystis giroveci causes intersti-tial pneumonia, which is pathogno-monic for T-cell deficiency;
b. Mucocutaneous infection with
Candida;
c. Systemic infections with filamentous fungi (e.g., Aspergillus fumigatus);
d. Meningitis or systemic infection caused by Cryptococcus neoformans.
4. Intracellular bacterial infection is a particular problem in T-cell-deficient patients. These patients are highly
susceptible to infection with M. tuberculosis or reactivation of latent tuberculosis. They are also suscep-tible to disseminated infections with poorly pathogenic mycobacteria (e.g., nontuberculous mycobacteria, BCG).
5. Infants with T-cell deficiency are usually lymphopenic and fail to thrive (i.e., fail to maintain normal rates of growth in height and weight).
6. Infants with SCID may develop der-matitis and hepatosplenomegaly due to graft-versus-host disease caused by maternal lymphocytes that have crossed the placenta.
7. Malignancies: T-cell-deficient indi-viduals are prone to develop a range of malignancies where viral infection acts as a co-factor (e.g., Epstein-Barr virus [EBV-] induced non-HodgkinŌĆÖs lymphoma, KaposiŌĆÖs sarcoma where human herpes virus 8 is a co-factor). There is also an increase in cutaneous malignancies occurring in an individ-ual exposed to significant amounts of ultraviolet light (basal-cell carcinoma and squamous carcinoma).
Major Categories of Combined Immunodeficiency
SEVERE COMBINED IMMUNODEFICIENCY
SCID comprises a group of inherited diseases characterized by a severe defi-cit in T-cell development and function with variable defects in B-cell and natural killer or NK cell development. SCID leads to death within the first two years of life, unless patients are rescued by hemato-poietic stem cell transplantation (HSCT). These are rare disorders with an estimated frequency between 1 in 50,000 and 1 in 100,000 live births.
Classification of SCID
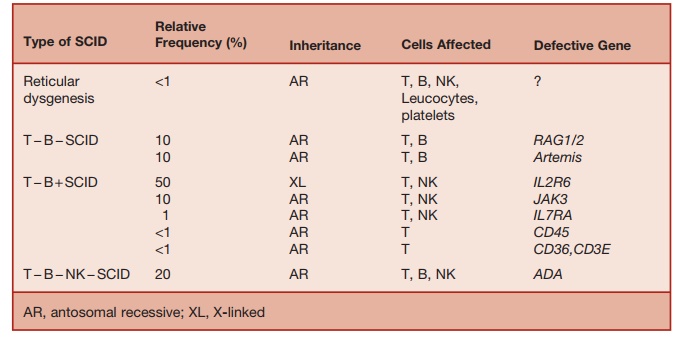
The classification of SCID, based on immunological and genetic criteria, is sum-marized in Table 5.6.
Table 5.6 Classification of SCID
SCID: Clinical Features
These patients typically present in the first year of life with failure to thrive and recurrent infections caused by bacterial, viral, and fungal pathogens. Infections typically affect the respiratory and gas-trointestinal systems. The infections may be caused by common pathogens (adenovirus, respiratory syncytial virus, parainfluenza virus), as well as by opportunistic organisms of low-grade virulence (Candida, Pneumocystis carinii, cytomegalovirus). Live vaccines such as BCG can lead to disseminated life-threat-ening infections. The persistent infections developing in SCID patients rapidly lead to malnutrition, growth impairment, and early death. Because of the patientŌĆÖs inability to reject allogeneic cells, graft-versus-host disease (GvHD) can be caused by transplacentally acquired maternal lymphocytes or by allogeneic cells following blood transfusion. GvHD manifests as skin rashes or hepatospleno-megaly and lymphadenopathy.
The absence of tonsils or other lymphatic tissue may be evident and radiographic studies may reveal thymic-hypoplasia. Lymphopenia (absolute lymphocyte count < 3 ├Ś 109/L in the first year of life) is a char-acteristic feature seen in over 80 percent of patients with SCID. (Therefore, SCID needs to be excluded in any infant with unexplained lymphopenia.)
Immunological and Molecular Classification of SCID
SCID can be classified in two groups based on the blood lymphocyte phenotype.
Ō¢Ā Patients lacking T cells with normal or increased B cells: TŌłÆB+ SCID
Ō¢Ā Patients lacking T and B cells: TŌłÆBŌłÆSCID
Defects in one of four functionally related genes causes TŌłÆB + SCID. X-linked SCID, which is the commonest form, is due to a mutation of the gene encod-ing the IL-2 receptor ╬│ chain, which is the signal-transducing chain common to the receptors for six cytokines (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21). The absence of responses to these cytokines causes defects in a broad range of T- and B-cell func-tions. IL-7 is required for early stages of T-cell development. Lack of response to this cytokine results in T lymphopenia. IL-15 is required for NK-cell development and its lack results in the failure of NK-cell development. Signal transduction through the aforementioned cytokine receptors involves the interaction of the common ╬│ chain with the tyrosine kinase JAK3. This explains why mutations of the JAK3 gene result in an autosomal recessive form of SCID, with a phenotype similar to X-linked SCID. Mutations of the ╬▒ chain of IL-2 or IL-7 receptors result in two rare forms of SCID.
T- and B-cell receptors consist of invari-ant signal-transducing elements combined with elements that make up the variable regions, which contribute to the antigen-binding portion of the receptor. The gene recombination required for generating these receptors requires the function of the product of recombination activating genes 1 and 2 and a number of proteins that are required for DNA repair (DNA-PKcs, KU70, KU80, XRLC4, and DNA-IV). In mice, mutations in any one of these genes produces analogues of SCID.
In humans, TŌłÆBŌłÆSCID is most common (50 percent of total) caused by mutations of the recombinase-activating genes, RAG1 or RAG2. RAG1 and RAG2 are enzymes responsible for introducing double-stranded DNA breaks, which initiate V(D)J gene rearrangements, required for gener-ating T- and B-cell receptors for antigen. Without normal RAG1 and RAG2 func-tion, T- and B-cell development is arrested early in ontogeny, producing TŌłÆBŌłÆSCID
Hypomorphic mutations of RAG1 or RAG2 result in a leaky form of SCID called OmennŌĆÖs syndrome. In OmennŌĆÖs syn-drome, a few T- and B-cell clones may be generated but the full T- and B-cell reper-toire fails to develop. The few T- and B-cell clones that leak through may undergo secondary expansion. As a result, patients with OmennŌĆÖs syndrome may not be mark-edly lymphopenic but the lymphocyte rep-ertoire is oligoclonal and severe immuno-deficiency is the outcome.
T- and B-cell antigen receptors are assembled from the recombination of variable region V(D)J and constant region genes. A protein called ARTEMIS is required for DNA repair, including the repair of DNA breaks generated during V(D)J recombination. Mutation of the gene encoding ARTEMIS results in a rare form of TŌłÆBŌłÆSCID. These patients also exhibit increased sensitivity to ionizing radiation.
About 15 percent of SCID cases are caused by deficiency of adenosine deami-nase (ADA), an enzyme required for the sal-vage of nucleotides within lymphoid cells. The lack of ADA causes the accumulation of toxic metabolites of adenosine (deoxy-adenosine and deoxy-ATP) within lym-phoid cells, resulting in their demise. ADA deficiency results in profound lymphopenia affecting T cells, B cells, and NK cells. Rarely, mutations of ADA causing milder forms of enzyme deficiency lead to a milder form of combined immunodeficiency presenting at a later stage in life. Purine nucleoside phos-phorylase (PNP) is an enzyme required for purine salvage within lymphocytes, and PNP deficiency causes a milder phenotype of SCID than seen in ADA deficiency. SCID due to PNP deficiency not treated with HSCT is fatal in childhood.
Mutations in proteins required for nor-mal functioning and signal transduction through the T-cell receptor (TCR) cause rare forms of SCID. Mutations of the tyro-sine phosphatase, CD45, which helps to initiate signaling by the TCR, results in TŌłÆB+ SCID in humans. Mutation of com-ponents of CD3-complex (CD3 ╬│, ╬Ą, and ╬┤) result in a SCID phenotype. During signal transduction via TCRs, the protein tyrosine kinases Lck and ZAP70 are required for phosphorylation of ITAMs on the intracy-toplasmic segment of the TCR. Deficiency of either of these kinases results in rare forms of SCID.
TCRs of CD8 cells recognize antigenic peptides that are complexed to MHC class I antigens, and TCR of CD4 cells recog-nize antigen bound to MHC class II on the surface of antigen-presenting cells. Cell-surface expression of MHC class I mol-ecules fails if either of the two transport-ers of antigenic peptides (TAP1 or TAP2) is lacking. TAP1 and TAP2 help to transfer peptides from the cytosol into the endo-plasmic reticulum, for subsequent loading onto newly synthesized MHC class I mol-ecules. In the absence of peptide loading, MHC class I molecules are degraded before reaching the cell surface. In the absence of MHC class I antigen expression, CD8 cell function is deficient, and these cells are not generated within the thymus. The result-ing immunodeficiency is milder than SCID and often presents in later life. Paradoxi-cally, viral infections are not a problem in these patients. Some MHC class I deficient patients develop progressive bronchiecta-sis, while others develop vasculitis affect-ing the face and upper respiratory tract. It has been postulated that vasculitis seen in these patients may be due to self-destruc-tion of vascular endothelial cells by the unrestrained cytotoxicity of NK cells.
In contrast, MHC class II deficiency results in a profound failure of CD4 cell
functions. Lack of thymic CD4+ CD8ŌłÆ cell selection for survival results in peripheral CD4 lymphopenia. Because CD4 func-tion is required for normal cell-mediated immunity, as well as antibody production, MHC class II deficiency results in a severe form of SCID with a fatal outcome. MHC class II deficiency is due to a mutation in one of four transcription factors (RFXAP, CIITA, RFX5, RFXANK), which regulate MHC class II expression.
TREATMENT OF SCID
Hematopoietic Stem Cell Transplantation
Untreated SCID is invariably fatal in early infancy. Once a diagnosis of SCID is confirmed, irrespective of the molecular diagnosis, HSCT from a human leucocyte antigen (HLA-) identical or haplo-identical family donor is the treatment of choice. Treatment of SCID with HSCT before 3.5 months of age results in good immune reconstitution and 95 percent survive long term. Delay in treatment, or the occurrence of infection, impairs outcome. Infection and GvHD are the main complications follow-ing HSCT. North American and European data indicate that long-term survival after transplants from HLA-matched unrelated donors was close to 60 percent. Review of European data between 1968 and 1999 indicates the progressive improvement of outcome, which is mainly because of better prevention of GvHD and the treatment of infection. Analysis of the outcome of European and U.S. bone marrow transplant programs for the treatment of SCID is ongoing and will be regularly reported.
Gene Therapy for SCID
Long-term immune reconstitution has been achieved in patients with SCID caused by the common ╬│-chain deficiency or ADA deficiency, using gene therapy.
This was achieved by ex vivo gene transfer to hematopoietic stem cells isolated from the patientŌĆÖs bone marrow. These gene-reconstituted stem cells were retransfused into the patient. To date, gene therapy has been restricted to patients without an HLA-matched family donor.
Several cases of leukemia have occurred among ╬│-chain-deficient patients who received gene therapy. In these cases, the retroviral vector had integrated close to the LMO2 proto-oncogene in the leukemia clone, leading to aberrant transcription and expression of LMO2. Because of this setback, clinical trials of gene therapy for SCID are being carefully evaluated, and more experience is required before the definitive role of gene therapy in SCID is established.
(<1 percent) exhibit profound T lympho-penia, associated with opportunistic infec-tions and a poor outlook unless rescued with fetal thymic transplant.
The cardiac, velopharyngeal, thymic, and parathyroid abnormalities are due to the defective development of third and fourth pharyngeal arches during ontogeny.
The 22q. 11.2 region contains the TBX1 gene, which belongs to the T-BOX fam-ily of genes that incorporate proteins that regulate embryonic development. Patients with mutations in the TBX1 genes also develop the clinical features seen in 22q. 11.2 deletion syndrome, suggesting that haplo-insufficiency of the TBX1 gene may be responsible for the clinical features seen in those with a deletion of the 22q. 11.2 region
DiGeorgeŌĆÖs Syndrome (Thymic Aplasia)
DiGeorgeŌĆÖs syndrome (DGS) is second-ary to a hemizygous deletion of the short arm of chromosome 22 (DEL 22q. 11.2). This chromosomal defect causes a com-plex inherited syndrome characterized by cardiac malformations, thymic hypopla-sia, palatopharyngeal abnormalities with associated velopharyngeal dysfunction, hypoparathyroidism, and facial dysmor-phism. The 22q deletion has an incidence of approximately 1 in 2,500 live births. The associated clinical phenotype is highly variable. About 20 percent of individu-als with 22q deletion have thymic aplasia, resulting in T lymphopenia and impaired CMI. In most such cases, the degree of T lymphopenia is modest (partial DGS) and almost complete restitution of the T-cell repertoire and function occurs by two years of age. Therefore, infections characteristic of T-cell deficiency are rare in these indi-viduals. A minority of infected individuals
Related Topics