Chapter: Essential Clinical Immunology: Immunological Aspects of Immunodeficiency Diseases
Defects in the Cytolytic Pathway
Defects in the Cytolytic Pathway
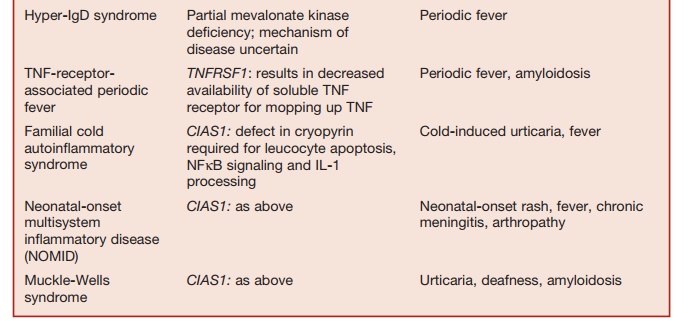
The homeostasis of immune responses requires prevention of excessive lymphocyte activation (Table 5.9). The mechanism by which such regulation occurs includes activation-induced cell death of T lymphocytes, which requires the activa- tion of apoptotic pathways. Defects in criti-cal components of these pathways result in susceptibility to hemophagocytic lym-phohistiocytosis (HLH), which is usually triggered by an intercurrent viral infection caused by viruses such as EBV or cytomeg-alovirus. HLH is characterized by massive infiltration of organs, such as the liver, spleen, bone marrow, and central nervous system, by activated CD8+ lymphocytes and macrophages, as well as a massive overproduction of IFN-γ and TNF-α. Severe pancytopenia is typical of this syndrome and is caused in part by phagocytosis of blood cells by activated macrophages and in part is secondary to the infiltration of bone marrow by activated macrophages (histiocytes).
A number of genetic defects that affect the efficiency of T-cell- and NK-cell-medi-ated cytolysis can predispose to the devel-opment of HLH. Cytolysis by T cells and NK cells is initiated by the secretion of contents of cytolytic granules at the immu-nological synapse between the T cells and the target cells. The process involves the translocation of perforin-containing lytic granules onto the target cell inter-phase, followed by fusion of these gran-ules with the plasma membrane of the T cell, with release of perforin onto the sur-face of the target cell. Perforin punches holes in the target cell membrane, causing cytolosis.
Mutation of the perforin (PRF1) gene, which encodes for perforin, is one of the genetic defects that predisposes to famil-ial HLH. Perforin-supported cytolysis (by CD-8 cells and NK cells) may damp down immune responses triggered by viral infections by aiding the elimination of antigen-presenting cells or by promoting activation-induced death of T cells.
Table 5.9 Disorders of Homeostasis of Immune Function
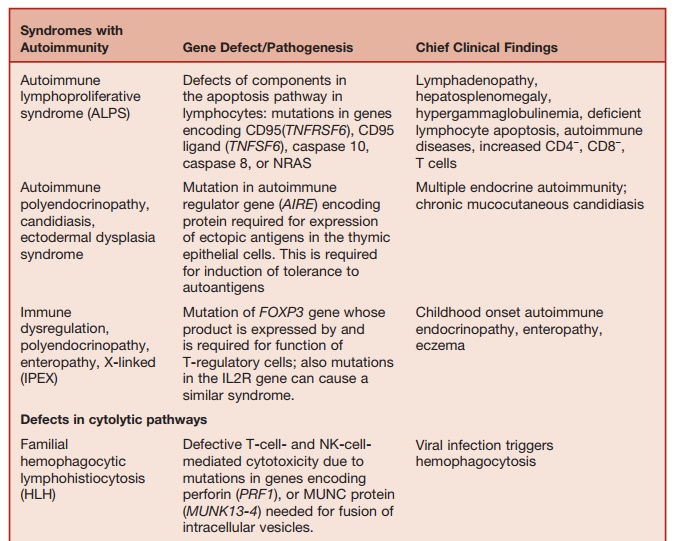
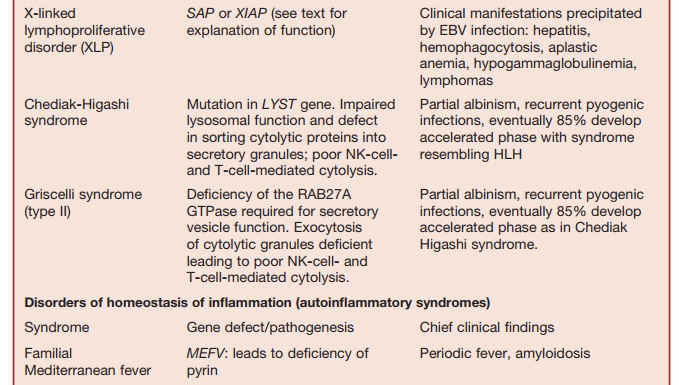
Studies of rare immunodeficiency syn-dromes, all of which are characterized by an increased tendency to develop HLH have identified a number of components required for the normal expression of cytolytic capacity by T cells and NK cells. The intracellular migration and docking of lytic granules requires the function of the small Rab GTPase, RAB27, which is mutated in the Griscelli syndrome char-acterized by partial albinism, immunode-ficiency, and susceptibility to developing HLH. Defective cytolytic granule exocy-tosis is also characteristic of patients with mutations of the gene encoding the protein MUNK13-4 (UNC13D). The tSNARE syn-texin 11, which is present in the trans-Golgi network, is also involved in intracellular vesicle trafficking. Mutation of these two genes is responsible for a further form of familial HLH.
Lysosomal trafficking regulator is defi-cient in individuals with a mutation in the CHS1 gene. This causes a defect in sorting of cytolytic proteins into secretory granules. This is the underlying defect in the condition called Chediak-Higashi syndrome, which is another condition characterized by susceptibility to HLH.
Related Topics